Non-Coding RNAs: Multi-Tasking Molecules in the Cell

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. The Incredible RNA Molecules
1.2. The Small Non-Coding RNAs
1.2.1. siRNAs and miRNAs
1.2.2. The piRNAs
2. Long Noncoding RNAs
3. The Emerging Roles of lncRNAS and miRNAs
3.1. LncRNAs: Implications in Different Levels of Gene Expression Regulation and Differentiation
3.1.1. Epigenetic Regulation
3.1.2. Transcriptional Regulation
3.1.3. Post-Transcriptional Regulation
3.1.4. Modulation of mRNA Nuclear Trafficking and Control of Nuclear Compartmentalization
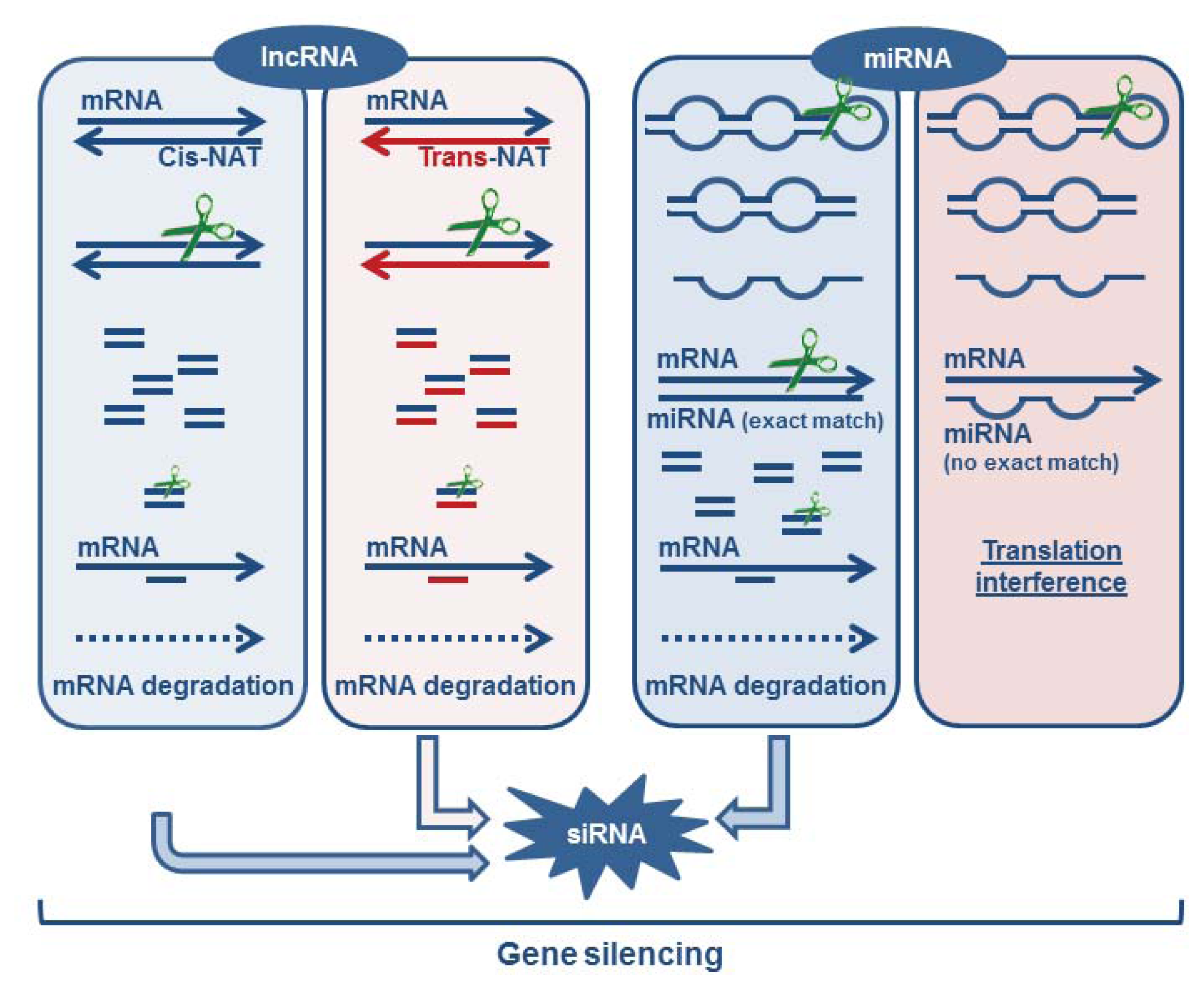
3.1.5. Formation of Endogenous siRNA
3.2. miRNAs as Critical Regulators of Target Degradation and Translation
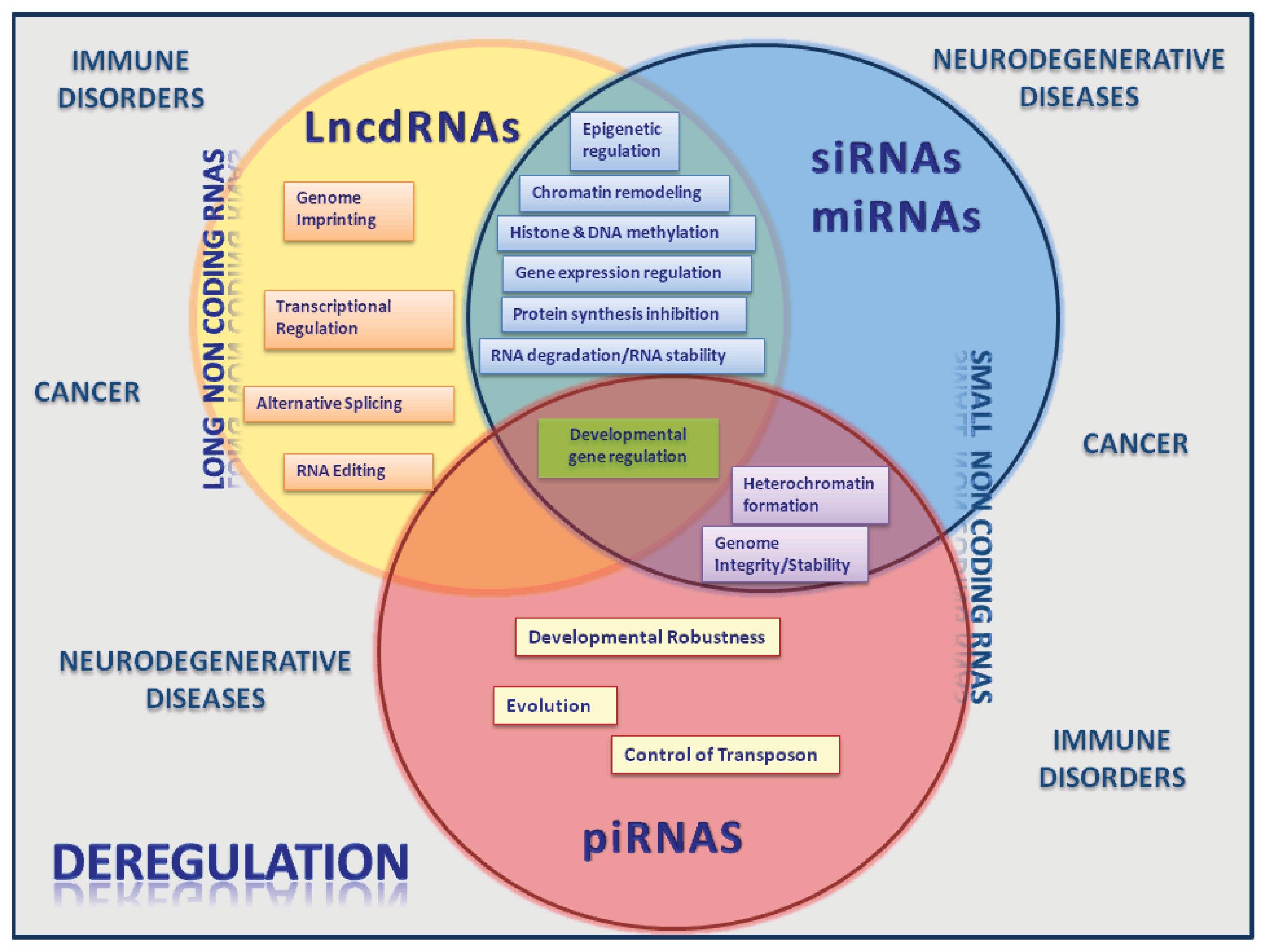
4. ncRNAs Active Players in Cancer and Other Human Diseases
5. Concluding Remarks
Acknowledgments
Conflict of Interest
References
- Cech, T.R.; Zaug, A.J.; Grabowski, P.J. In vitro splicing of the ribosomal RNA precursor of Tetrahymena: Involvement of a guanosine nucleotide in the excision of the intervening sequence. Cell 1981, 27, 487–496. [Google Scholar]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar]
- Dworkin, J.P.; Lazcano, A.; Miller, S.L. The roads to and from the RNA world. J. Theor. Boil 2003, 222, 127–134. [Google Scholar]
- Lehman, N. RNA in evolution. Wiley Interdiscip. Rev 2010, 1, 202–213. [Google Scholar]
- Noller, H.F.; Hoffarth, V.; Zimniak, L. Unusual resistance of peptidyl transferase to protein extraction procedures. Science 1992, 256, 1416–1419. [Google Scholar]
- Von Ahsen, U.; Noller, H.F. Identification of bases in 16S rRNA essential for tRNA binding at the 30S ribosomal P site. Science 1995, 267, 234–237. [Google Scholar]
- Moazed, D.; Noller, H.F. Sites of interaction of the CCA end of peptidyl-tRNA with 23S rRNA. Proc. Natl. Acad. Sci. USA 1991, 88, 3725–3728. [Google Scholar]
- Wassarman, D.A.; Steitz, J.A. Interactions of small nuclear RNA’s with precursor messenger RNA during in vitro splicing. Science 1992, 257, 1918–1925. [Google Scholar]
- Brennicke, A.; Marchfelder, A.; Binder, S. RNA editing. FEMS Microbial. Rev 1999, 23, 297–316. [Google Scholar]
- Bachellerie, J.P.; Cavaille, J.; Huttenhofer, A. The expanding snoRNA world. Biochimie 2002, 84, 775–790. [Google Scholar]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar]
- Qiao, F.; Cech, T.R. Triple-helix structure in telomerase RNA contributes to catalysis. Nat. Struct. Mol. Boil 2008, 15, 634–640. [Google Scholar]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigo, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet 2006, 15, R17–R29. [Google Scholar]
- Moazed, D. Small RNAs in transcriptional gene silencing and genome defence. Nature 2009, 457, 413–420. [Google Scholar]
- Eliceiri, G.L. Small nucleolar RNAs. Cell. Mol. Life Sci 1999, 56, 22–31. [Google Scholar]
- Mannoor, K.; Liao, J.; Jiang, F. Small nucleolar RNAs in cancer. Biochim. Biophys. Acta 2012, 1826, 121–128. [Google Scholar]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar]
- Jinek, M.; Doudna, J.A. A three-dimensional view of the molecular machinery of RNA interference. Nature 2009, 457, 405–412. [Google Scholar]
- Michlewski, G.; Guil, S.; Semple, C.A.; Cáceres, J.F. Posttranscriptional regulation of miRNAs harboring conserved terminal loops. Mol. Cell 2008, 7, 383–393. [Google Scholar]
- Zeng, Y.; Cullen, B.R. Structural requirements for pre-microRNA binding and nuclear export by Exportin 5. Nucleic Acids Res 2004, 32, 4776–4785. [Google Scholar]
- Berezikov, E.; Chung, W.J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian mirtron genes. Mol. Cell 2007, 28, 328–336. [Google Scholar]
- Farazi, T.A.; Juranek, S.A.; Tuschl, T. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development 2008, 135, 1201–1214. [Google Scholar]
- Forstemann, K.; Horwich, M.D.; Wee, L.; Tomari, Y.; Zamore, P.D. Drosophila microRNAs are sorted into functionally distinct argonaute complexes after production by dicer-1. Cell 2007, 130, 287–297. [Google Scholar]
- Tomari, Y.; Du, T.; Zamore, P.D. Sorting of Drosophila small silencing RNAs. Cell 2007, 130, 299–308. [Google Scholar]
- Ameres, S.L.; Horwich, M.D.; Hung, J.H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar]
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.J.; Park, J.E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 2012, 151, 521–532. [Google Scholar]
- Khvorova, A.; Reynolds, A.; Jayasena, S.D. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003, 115, 209–216. [Google Scholar]
- Siomi, H.; Siomi, M.C. On the road to reading the RNA-interference code. Nature 2009, 457, 396–404. [Google Scholar]
- Fukunaga, R.; Han, B.W.; Hung, J.H.; Xu, J.; Weng, Z.; Zamore, P.D. Dicer partner proteins tune the length of mature miRNAs in flies and mammals. Cell 2012, 151, 533–546. [Google Scholar]
- Lee, H.Y.; Doudna, J.A. TRBP alters human precursor microRNA processing in vitro. RNA 2012, 18, 2012–2019. [Google Scholar]
- Grewal, S.I.; Elgin, S.C. Transcription and RNA interference in the formation of heterochromatin. Nature 2007, 447, 399–406. [Google Scholar]
- Verdel, A.; Jia, S.; Gerber, S.; Sugiyama, T.; Gygi, S.; Grewal, S.I.; Moazed, D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science 2004, 303, 672–676. [Google Scholar]
- Creamer, K.M.; Partridge, J.F. RITS-connecting transcription, RNA interference, and heterochromatin assembly in fission yeast. Wiley Interdiscip. Rev 2011, 2, 632–646. [Google Scholar]
- Aravin, A.A.; Lagos-Quintana, M.; Yalcin, A.; Zavolan, M.; Marks, D.; Snyder, B.; Gaasterland, T.; Meyer, J.; Tuschl, T. The small RNA profile during Drosophila melanogaster development. Dev. cell 2003, 5, 337–350. [Google Scholar]
- Lau, N.C.; Seto, A.G.; Kim, J.; Kuramochi-Miyagawa, S.; Nakano, T.; Bartel, D.P.; Kingston, R.E. Characterization of the piRNA complex from rat testes. Science 2006, 313, 363–367. [Google Scholar]
- Aravin, A.A.; Hannon, G.J.; Brennecke, J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 2007, 318, 761–764. [Google Scholar]
- Brennecke, J.; Aravin, A.A.; Stark, A.; Dus, M.; Kellis, M.; Sachidanandam, R.; Hannon, G.J. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 2007, 128, 1089–1103. [Google Scholar]
- Houwing, S.; Kamminga, L.M.; Berezikov, E.; Cronembold, D.; Girard, A.; van den Elst, H.; Filippov, D.V.; Blaser, H.; Raz, E.; Moens, C.B.; et al. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 2007, 129, 69–82. [Google Scholar]
- Robine, N.; Lau, N.C.; Balla, S.; Jin, Z.; Okamura, K.; Kuramochi-Miyagawa, S.; Blower, M.D.; Lai, E.C. A broadly conserved pathway generates 3′ UTR-directed primary piRNAs. Curr. Biol 2009, 19, 2066–2076. [Google Scholar]
- Khurana, J.S.; Theurkauf, W. piRNAs, transposon silencing, and Drosophila germline development. J. Cell Biol 2010, 191, 905–913. [Google Scholar]
- Chen, Y.; Pane, A.; Schupbach, T. Cutoff and aubergine mutations result in retrotransposon upregulation and checkpoint activation in Drosophila. Curr. Biol 2007, 17, 637–642. [Google Scholar]
- Khurana, J.S.; Xu, J.; Weng, Z.; Theurkauf, W.E. Distinct functions for the Drosophila piRNA pathway in genome maintenance and telomere protection. PLoS Genet 2010, 6, e1001246. [Google Scholar]
- Gunawardane, L.S.; Saito, K.; Nishida, K.M.; Miyoshi, K.; Kawamura, Y.; Nagami, T.; Siomi, H.; Siomi, M.C. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science 2007, 315, 1587–1590. [Google Scholar]
- Ishizu, H.; Siomi, H.; Siomi, M.C. Biology of PIWI-interacting RNAs: New insights into biogenesis and function inside and outside of germlines. Genes Dev 2012, 26, 2361–2373. [Google Scholar]
- Yan, B.X.; Ma, J.X. Promoter-associated RNAs and promoter-targeted RNAs. Cell. Mol. Life Sci 2012, 69, 2833–2842. [Google Scholar]
- Rouget, C.; Papin, C.; Boureux, A.; Meunier, A.C.; Franco, B.; Robine, N.; Lai, E.C.; Pelisson, A.; Simonelig, M. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature 2010, 467, 1128–1132. [Google Scholar]
- Lee, E.J.; Banerjee, S.; Zhou, H.; Jammalamadaka, A.; Arcila, M.; Manjunath, B.S.; Kosik, K.S. Identification of piRNAs in the central nervous system. RNA 2011, 17, 1090–1099. [Google Scholar]
- Rajasethupathy, P.; Antonov, I.; Sheridan, R.; Frey, S.; Sander, C.; Tuschl, T.; Kandel, E.R. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 2012, 149, 693–707. [Google Scholar]
- Sienski, G.; Donertas, D.; Brennecke, J. Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 2012, 151, 964–980. [Google Scholar]
- Watanabe, T.; Tomizawa, S.; Mitsuya, K.; Totoki, Y.; Yamamoto, Y.; Kuramochi-Miyagawa, S.; Iida, N.; Hoki, Y.; Murphy, P.J.; Toyoda, A.; et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 2011, 332, 848–852. [Google Scholar]
- Gangaraju, V.K.; Yin, H.; Weiner, M.M.; Wang, J.; Huang, X.A.; Lin, H. Drosophila Piwi functions in Hsp90-mediated suppression of phenotypic variation. Nat. genet 2011, 43, 153–158. [Google Scholar]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar]
- Specchia, V.; Piacentini, L.; Tritto, P.; Fanti, L.; D’Alessandro, R.; Palumbo, G.; Pimpinelli, S.; Bozzetti, M.P. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature 2010, 463, 662–665. [Google Scholar]
- Kim, V.N. Small RNAs just got bigger: Piwi-interacting RNAs (piRNAs) in mammalian testes. Genes Dev 2006, 20, 1993–1997. [Google Scholar]
- Siddiqi, S.; Matushansky, I. Piwis and piwi-interacting RNAs in the epigenetics of cancer. J. Cell. Biochem 2012, 113, 373–380. [Google Scholar]
- Siddiqi, S.; Terry, M.; Matushansky, I. Hiwi mediated tumorigenesis is associated with DNA hypermethylation. PLoS One 2012, 7, e33711. [Google Scholar]
- Prescott, D.M. The DNA of ciliated protozoa. Microbiol. Rev. 1994, 58, 233–267. [Google Scholar]
- Chalker, D.L. Dynamic nuclear reorganization during genome remodeling of Tetrahymena. Biochim. Biophys. Acta 2008, 1783, 2130–2136. [Google Scholar]
- Duharcourt, S.; Lepere, G.; Meyer, E. Developmental genome rearrangements in ciliates: A natural genomic subtraction mediated by non-coding transcripts. Trends Genet 2009, 25, 344–350. [Google Scholar]
- Lee, J.T. Epigenetic regulation by long noncoding RNAs. Science 2012, 338, 1435–1439. [Google Scholar]
- Pink, R.C.; Wicks, K.; Caley, D.P.; Punch, E.K.; Jacobs, L.; Carter, D.R. Pseudogenes: Pseudo-functional or key regulators in health and disease? RNA 2011, 17, 792–798. [Google Scholar]
- Rackham, O.; Shearwood, A.M.; Mercer, T.R.; Davies, S.M.; Mattick, J.S.; Filipovska, A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA 2011, 17, 2085–2093. [Google Scholar]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar]
- Magistri, M.; Faghihi, M.A.; St Laurent, G., 3rd; Wahlestedt, C. Regulation of chromatin structure by long noncoding RNAs: Focus on natural antisense transcripts. Trends Genet. 2012, 28, 389–396. [Google Scholar]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar]
- Orom, U.A.; Derrien, T.; Beringer, M.; Gumireddy, K.; Gardini, A.; Bussotti, G.; Lai, F.; Zytnicki, M.; Notredame, C.; Huang, Q.; et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010, 143, 46–58. [Google Scholar]
- Kim, T.K.; Hemberg, M.; Gray, J.M.; Costa, A.M.; Bear, D.M.; Wu, J.; Harmin, D.A.; Laptewicz, M.; Barbara-Haley, K.; Kuersten, S.; et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010, 465, 182–187. [Google Scholar]
- Katayama, S.; Tomaru, Y.; Kasukawa, T.; Waki, K.; Nakanishi, M.; Nakamura, M.; Nishida, H.; Yap, C.C.; Suzuki, M.; Kawai, J.; et al. Antisense transcription in the mammalian transcriptome. Science 2005, 309, 1564–1566. [Google Scholar]
- Korneev, S.A.; Korneeva, E.I.; Lagarkova, M.A.; Kiselev, S.L.; Critchley, G.; O’Shea, M. Novel noncoding antisense RNA transcribed from human anti-NOS2A locus is differentially regulated during neuronal differentiation of embryonic stem cells. RNA 2008, 14, 2030–2037. [Google Scholar]
- Lavorgna, G.; Dahary, D.; Lehner, B.; Sorek, R.; Sanderson, C.M.; Casari, G. In search of antisense. Trends Biochem. Sci 2004, 29, 88–94. [Google Scholar]
- Werner, A.; Carlile, M.; Swan, D. What do natural antisense transcripts regulate? RNA Biol 2009, 6, 43–48. [Google Scholar]
- He, Y.; Vogelstein, B.; Velculescu, V.E.; Papadopoulos, N.; Kinzler, K.W. The antisense transcriptomes of human cells. Science 2008, 322, 1855–1857. [Google Scholar]
- Ravasi, T.; Suzuki, H.; Pang, K.C.; Katayama, S.; Furuno, M.; Okunishi, R.; Fukuda, S.; Ru, K.; Frith, M.C.; Gongora, M.M.; et al. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res 2006, 16, 11–19. [Google Scholar]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.J.; Lee, J.T. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 2008, 322, 750–756. [Google Scholar]
- Cai, X.; Cullen, B.R. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 2007, 13, 313–316. [Google Scholar]
- Hung, T.; Chang, H.Y. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol 2010, 7, 582–585. [Google Scholar]
- Mohammad, F.; Mondal, T.; Guseva, N.; Pandey, G.K.; Kanduri, C. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 2010, 137, 2493–2499. [Google Scholar]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Swiezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold-induced silencing by long antisense transcripts of an Arabidopsis polycomb target. Nature 2009, 462, 799–802. [Google Scholar]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2011, 331, 76–79. [Google Scholar]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet 2011, 43, 621–629. [Google Scholar]
- Bertani, S.; Sauer, S.; Bolotin, E.; Sauer, F. The noncoding RNA Mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting MLL1 to chromatin. Mol. Cell 2011, 43, 1040–1046. [Google Scholar]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar]
- Dinger, M.E.; Amaral, P.P.; Mercer, T.R.; Pang, K.C.; Bruce, S.J.; Gardiner, B.B.; Askarian-Amiri, M.E.; Ru, K.; Solda, G.; Simons, C.; et al. Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res 2008, 18, 1433–1445. [Google Scholar]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011, 25, 1915–1927. [Google Scholar]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol 2013, 20, 300–307. [Google Scholar]
- Su, W.Y.; Xiong, H.; Fang, J.Y. Natural antisense transcripts regulate gene expression in an epigenetic manner. Biochem. Biophys. Res. Commun 2010, 396, 177–181. [Google Scholar]
- Imamura, T.; Yamamoto, S.; Ohgane, J.; Hattori, N.; Tanaka, S.; Shiota, K. Non-coding RNA directed DNA demethylation of Sphk1 CpG island. Biochem. Biophys. Res. Commun 2004, 322, 593–600. [Google Scholar]
- Berretta, J.; Pinskaya, M.; Morillon, A. A cryptic unstable transcript mediates transcriptional trans-silencing of the Ty1 retrotransposon in S. cerevisiae. Genes Dev 2008, 22, 615–626. [Google Scholar]
- Martianov, I.; Ramadass, A.; Serra Barros, A.; Chow, N.; Akoulitchev, A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 2007, 445, 666–670. [Google Scholar]
- Morris, K.V.; Chan, S.W.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar]
- Napoli, S.; Pastori, C.; Magistri, M.; Carbone, G.M.; Catapano, C.V. Promoter-specific transcriptional interference and c-myc gene silencing by siRNAs in human cells. EMBO J 2009, 28, 1708–1719. [Google Scholar]
- Hawkins, P.G.; Santoso, S.; Adams, C.; Anest, V.; Morris, K.V. Promoter targeted small RNAs induce long-term transcriptional gene silencing in human cells. Nucleic Acids Res 2009, 37, 2984–2995. [Google Scholar]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. microRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar]
- Li, L.C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar]
- Janowski, B.A.; Younger, S.T.; Hardy, D.B.; Ram, R.; Huffman, K.E.; Corey, D.R. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat. Chem. Biol 2007, 3, 166–173. [Google Scholar]
- Natoli, G.; Andrau, J.C. Noncoding transcription at enhancers: General principles and functional models. Annu. Rev. Genet 2012, 46, 1–19. [Google Scholar]
- Orom, U.A.; Shiekhattar, R. Long non-coding RNAs and enhancers. Curr. Opin. Genet. Dev 2011, 21, 194–198. [Google Scholar]
- Panganiban, G.; Rubenstein, J.L. Developmental functions of the Distal-less/Dlx homeobox genes. Development 2002, 129, 4371–4386. [Google Scholar]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar]
- Goodrich, J.A.; Kugel, J.F. Non-coding-RNA regulators of RNA polymerase II transcription. Nat. Rev 2006, 7, 612–616. [Google Scholar]
- Hastings, M.L.; Ingle, H.A.; Lazar, M.A.; Munroe, S.H. Post-transcriptional regulation of thyroid hormone receptor expression by cis-acting sequences and a naturally occurring antisense RNA. J. Boil. Chem 2000, 275, 11507–11513. [Google Scholar]
- Yin, Q.F.; Yang, L.; Zhang, Y.; Xiang, J.F.; Wu, Y.W.; Carmichael, G.G.; Chen, L.L. Long noncoding RNAs with snoRNA ends. Mol. Cell 2012, 48, 219–230. [Google Scholar]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med 2008, 14, 723–730. [Google Scholar]
- Ebralidze, A.K.; Guibal, F.C.; Steidl, U.; Zhang, P.; Lee, S.; Bartholdy, B.; Jorda, M.A.; Petkova, V.; Rosenbauer, F.; Huang, G.; et al. PU.1 expression is modulated by the balance of functional sense and antisense RNAs regulated by a shared cis-regulatory element. Genes Dev 2008, 22, 2085–2092. [Google Scholar]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science 2005, 309, 1570–1573. [Google Scholar]
- Guo, X.; Zhang, Z.; Gerstein, M.B.; Zheng, D. Small RNAs originated from pseudogenes: cis- or trans-acting? PLoS Comput. Biol 2009, 5, e1000449. [Google Scholar]
- Tam, O.H.; Aravin, A.A.; Stein, P.; Girard, A.; Murchison, E.P.; Cheloufi, S.; Hodges, E.; Anger, M.; Sachidanandam, R.; Schultz, R.M.; et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 2008, 453, 534–538. [Google Scholar]
- Watanabe, T.; Totoki, Y.; Toyoda, A.; Kaneda, M.; Kuramochi-Miyagawa, S.; Obata, Y.; Chiba, H.; Kohara, Y.; Kono, T.; Nakano, T.; et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 2008, 453, 539–543. [Google Scholar]
- Mighell, A.J.; Smith, N.R.; Robinson, P.A.; Markham, A.F. Vertebrate pseudogenes. FEBS Lett 2000, 468, 109–114. [Google Scholar]
- Zhang, Z.D.; Frankish, A.; Hunt, T.; Harrow, J.; Gerstein, M. Identification and analysis of unitary pseudogenes: Historic and contemporary gene losses in humans and other primates. Genome Biol 2010, 11, R26. [Google Scholar]
- Maestre, J.; Tchenio, T.; Dhellin, O.; Heidmann, T. mRNA retroposition in human cells: Processed pseudogene formation. EMBO J 1995, 14, 6333–6338. [Google Scholar]
- D’Errico, I.; Gadaleta, G.; Saccone, C. Pseudogenes in metazoa: Origin and features. Briefings Funct. Genomics Proteomics 2004, 3, 157–167. [Google Scholar]
- Werner, A.; Schmutzler, G.; Carlile, M.; Miles, C.G.; Peters, H. Expression profiling of antisense transcripts on DNA arrays. Physiol. Genomics 2007, 28, 294–300. [Google Scholar]
- Fanarraga, M.L.; Parraga, M.; Aloria, K.; del Mazo, J.; Avila, J.; Zabala, J.C. Regulated expression of p14 (cofactor A) during spermatogenesis. Cell Motil. Cytoskeleton 1999, 43, 243–254. [Google Scholar]
- Pavlicek, A.; Gentles, A.J.; Paces, J.; Paces, V.; Jurka, J. Retroposition of processed pseudogenes: The impact of RNA stability and translational control. Trends Genet 2006, 22, 69–73. [Google Scholar]
- Borsani, O.; Zhu, J.; Verslues, P.E.; Sunkar, R.; Zhu, J.K. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 2005, 123, 1279–1291. [Google Scholar]
- Okada, Y.; Tashiro, C.; Numata, K.; Watanabe, K.; Nakaoka, H.; Yamamoto, N.; Okubo, K.; Ikeda, R.; Saito, R.; Kanai, A.; et al. Comparative expression analysis uncovers novel features of endogenous antisense transcription. Hum. Mol. Genet 2008, 17, 1631–1640. [Google Scholar]
- Okamoto, I.; Arnaud, D.; le Baccon, P.; Otte, A.P.; Disteche, C.M.; Avner, P.; Heard, E. Evidence for de novo imprinted X-chromosome inactivation independent of meiotic inactivation in mice. Nature 2005, 438, 369–373. [Google Scholar]
- Young, T.L.; Matsuda, T.; Cepko, C.L. The noncoding RNA taurine upregulated gene 1 is required for differentiation of the murine retina. Curr. Biol 2005, 15, 501–512. [Google Scholar]
- Ginger, M.R.; Shore, A.N.; Contreras, A.; Rijnkels, M.; Miller, J.; Gonzalez-Rimbau, M.F.; Rosen, J.M. A noncoding RNA is a potential marker of cell fate during mammary gland development. Proc. Natl. Acad. Sci. USA 2006, 103, 5781–5786. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar]
- Loewer, S.; Cabili, M.N.; Guttman, M.; Loh, Y.H.; Thomas, K.; Park, I.H.; Garber, M.; Curran, M.; Onder, T.; Agarwal, S.; et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet 2010, 42, 1113–1117. [Google Scholar] [Green Version]
- Kierszenbaum, A.L.; Tres, L.L. The acrosome-acroplaxome-manchette complex and the shaping of the spermatid head. Arch. Histol. Cytol 2004, 67, 271–284. [Google Scholar]
- Nolasco, S.; Bellido, J.; Goncalves, J.; Tavares, A.; Zabala, J.C.; Soares, H. The expression of tubulin cofactor A (TBCA) is regulated by a noncoding antisense Tbca RNA during testis maturation. PLoS One 2012, 7, e42536. [Google Scholar]
- Bartel, D.P. microRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar]
- Jing, Q.; Huang, S.; Guth, S.; Zarubin, T.; Motoyama, A.; Chen, J.; di Padova, F.; Lin, S.C.; Gram, H.; Han, J. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell 2005, 120, 623–634. [Google Scholar]
- Behm-Ansmant, I.; Rehwinkel, J.; Doerks, T.; Stark, A.; Bork, P.; Izaurralde, E. mRNA degradation by miRNAs and GW182 requires both CCR4: NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev 2006, 20, 1885–1898. [Google Scholar]
- Llave, C.; Xie, Z.; Kasschau, K.D.; Carrington, J.C. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 2002, 297, 2053–2056. [Google Scholar]
- Kim, D.H.; Saetrom, P.; Snove, O., Jr; Rossi, J.J. microRNA-directed transcriptional gene silencing in mammalian cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16230–16235. [Google Scholar]
- Liu, J.; Valencia-Sanchez, M.A.; Hannon, G.J.; Parker, R. microRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol 2005, 7, 719–723. [Google Scholar]
- Pillai, R.S.; Bhattacharyya, S.N.; Artus, C.G.; Zoller, T.; Cougot, N.; Basyuk, E.; Bertrand, E.; Filipowicz, W. Inhibition of translational initiation by Let-7 microRNA in human cells. Science 2005, 309, 1573–1576. [Google Scholar]
- Humphreys, D.T.; Westman, B.J.; Martin, D.I.; Preiss, T. microRNAs control translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(A) tail function. Proc. Natl. Acad. Sci. USA 2005, 102, 16961–16966. [Google Scholar]
- Kiriakidou, M.; Tan, G.S.; Lamprinaki, S.; de Planell-Saguer, M.; Nelson, P.T.; Mourelatos, Z. An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell 2007, 129, 1141–1151. [Google Scholar]
- Eulalio, A.; Huntzinger, E.; Izaurralde, E. GW182 interaction with Argonaute is essential for miRNA-mediated translational repression and mRNA decay. Nat. Struct. Mol. Biol 2008, 15, 346–353. [Google Scholar]
- Wang, B.; Yanez, A.; Novina, C.D. microRNA-repressed mRNAs contain 40S but not 60S components. Proc. Natl. Acad. Sci. USA 2008, 105, 5343–5348. [Google Scholar]
- Thermann, R.; Hentze, M.W. Drosophila miR2 induces pseudo-polysomes and inhibits translation initiation. Nature 2007, 447, 875–878. [Google Scholar]
- Maroney, P.A.; Yu, Y.; Fisher, J.; Nilsen, T.W. Evidence that microRNAs are associated with translating messenger RNAs in human cells. Nat. Struct. Mol. Biol 2006, 13, 1102–1107. [Google Scholar]
- Petersen, C.P.; Bordeleau, M.E.; Pelletier, J.; Sharp, P.A. Short RNAs repress translation after initiation in mammalian cells. Mol. Cell 2006, 21, 533–542. [Google Scholar]
- Lytle, J.R.; Yario, T.A.; Steitz, J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc. Natl. Acad. Sci. USA 2007, 104, 9667–9672. [Google Scholar]
- Vasudevan, S.; Steitz, J.A. AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 2007, 128, 1105–1118. [Google Scholar]
- Orom, U.A.; Nielsen, F.C.; Lund, A.H. microRNA-10a binds the 5′ UTR of ribosomal protein mRNAs and enhances their translation. Mol. Cell 2008, 30, 460–471. [Google Scholar]
- Lee, S.; Vasudevan, S. Post-transcriptional stimulation of gene expression by microRNAs. Adv. Exper. Med. Biol 2013, 768, 97–126. [Google Scholar]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet 2003, 35, 215–217. [Google Scholar]
- Leonardo, T.R.; Schultheisz, H.L.; Loring, J.F.; Laurent, L.C. The functions of microRNAs in pluripotency and reprogramming. Nat. Cell Biol 2012, 14, 1114–1121. [Google Scholar]
- Giraldez, A.J.; Cinalli, R.M.; Glasner, M.E.; Enright, A.J.; Thomson, J.M.; Baskerville, S.; Hammond, S.M.; Bartel, D.P.; Schier, A.F. microRNAs regulate brain morphogenesis in zebrafish. Science 2005, 308, 833–838. [Google Scholar]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet 2008, 40, 751–760. [Google Scholar]
- Cobb, B.S.; Nesterova, T.B.; Thompson, E.; Hertweck, A.; O’Connor, E.; Godwin, J.; Wilson, C.B.; Brockdorff, N.; Fisher, A.G.; Smale, S.T.; et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J. Exp. Med 2005, 201, 1367–1373. [Google Scholar]
- Muljo, S.A.; Ansel, K.M.; Kanellopoulou, C.; Livingston, D.M.; Rao, A.; Rajewsky, K. Aberrant T cell differentiation in the absence of Dicer. J. Exp. Med 2005, 202, 261–269. [Google Scholar]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar]
- Monticelli, S.; Ansel, K.M.; Xiao, C.; Socci, N.D.; Krichevsky, A.M.; Thai, T.H.; Rajewsky, N.; Marks, D.S.; Sander, C.; Rajewsky, K.; et al. microRNA profiling of the murine hematopoietic system. Genome Biol 2005, 6, R71. [Google Scholar]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med 2011, 208, 1189–1201. [Google Scholar]
- Steiner, D.F.; Thomas, M.F.; Hu, J.K.; Yang, Z.; Babiarz, J.E.; Allen, C.D.; Matloubian, M.; Blelloch, R.; Ansel, K.M. microRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity 2011, 35, 169–181. [Google Scholar]
- Cheetham, S.W.; Gruhl, F.; Mattick, J.S.; Dinger, M.E. Long noncoding RNAs and the genetics of cancer. Br. J. Cancer 2013, 108, 2419–2425. [Google Scholar]
- Munker, R.; Calin, G.A. microRNA profiling in cancer. Clin. Sci 2011, 121, 141–158. [Google Scholar]
- Faraoni, I.; Antonetti, F.R.; Cardone, J.; Bonmassar, E. miR-155 gene: A typical multifunctional microRNA. Biochim. Biophys. Acta 2009, 1792, 497–505. [Google Scholar]
- Chan, J.A.; Krichevsky, A.M.; Kosik, K.S. microRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005, 65, 6029–6033. [Google Scholar]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar]
- Xiao, C.; Srinivasan, L.; Calado, D.P.; Patterson, H.C.; Zhang, B.; Wang, J.; Henderson, J.M.; Kutok, J.L.; Rajewsky, K. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat. Immunol 2008, 9, 405–414. [Google Scholar]
- Concepcion, C.P.; Bonetti, C.; Ventura, A. The microRNA-17–92 family of microRNA clusters in development and disease. Cancer J 2012, 18, 262–267. [Google Scholar]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar]
- Gutschner, T.; Hammerle, M.; Eissmann, M.; Hsu, J.; Kim, Y.; Hung, G.; Revenko, A.; Arun, G.; Stentrup, M.; Gross, M.; et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 2013, 73, 1180–1189. [Google Scholar]
- Drakaki, A.; Iliopoulos, D. microRNA gene networks in oncogenesis. Curr. Genomics 2009, 10, 35–41. [Google Scholar]
- Dalmay, T.; Edwards, D.R. microRNAs and the hallmarks of cancer. Oncogene 2006, 25, 6170–6175. [Google Scholar]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol 2012, 9, 703–719. [Google Scholar]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar]
- Concepcion, C.P.; Han, Y.C.; Mu, P.; Bonetti, C.; Yao, E.; D’Andrea, A.; Vidigal, J.A.; Maughan, W.P.; Ogrodowski, P.; Ventura, A. Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet 2012, 8, e1002797. [Google Scholar]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res 2010, 38, 5797–5806. [Google Scholar]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci 2008, 28, 14341–14346. [Google Scholar]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J. Alzheimer’s Disease 2008, 14, 27–41. [Google Scholar]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A microRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar]
- Contreras, J.; Rao, D.S. microRNAs in inflammation and immune responses. Leukemia 2012, 26, 404–413. [Google Scholar]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 2012, 149, 819–831. [Google Scholar]
- Szafranski, P.; Dharmadhikari, A.V.; Brosens, E.; Gurha, P.; Kolodziejska, K.E.; Zhishuo, O.; Dittwald, P.; Majewski, T.; Mohan, K.N.; Chen, B.; et al. Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res 2013, 23, 23–33. [Google Scholar]
- Van Dijk, M.; Thulluru, H.K.; Mulders, J.; Michel, O.J.; Poutsma, A.; Windhorst, S.; Kleiverda, G.; Sie, D.; Lachmeijer, A.M.; Oudejans, C.B. HELLP babies link a novel lincRNA to the trophoblast cell cycle. J. Clin. Invest 2012, 122, 4003–4011. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gomes, A.Q.; Nolasco, S.; Soares, H. Non-Coding RNAs: Multi-Tasking Molecules in the Cell. Int. J. Mol. Sci. 2013, 14, 16010-16039. https://doi.org/10.3390/ijms140816010
Gomes AQ, Nolasco S, Soares H. Non-Coding RNAs: Multi-Tasking Molecules in the Cell. International Journal of Molecular Sciences. 2013; 14(8):16010-16039. https://doi.org/10.3390/ijms140816010
Chicago/Turabian StyleGomes, Anita Quintal, Sofia Nolasco, and Helena Soares. 2013. "Non-Coding RNAs: Multi-Tasking Molecules in the Cell" International Journal of Molecular Sciences 14, no. 8: 16010-16039. https://doi.org/10.3390/ijms140816010