Oxidative Stress and MicroRNAs in Vascular Diseases


Abstract
:1. Introduction
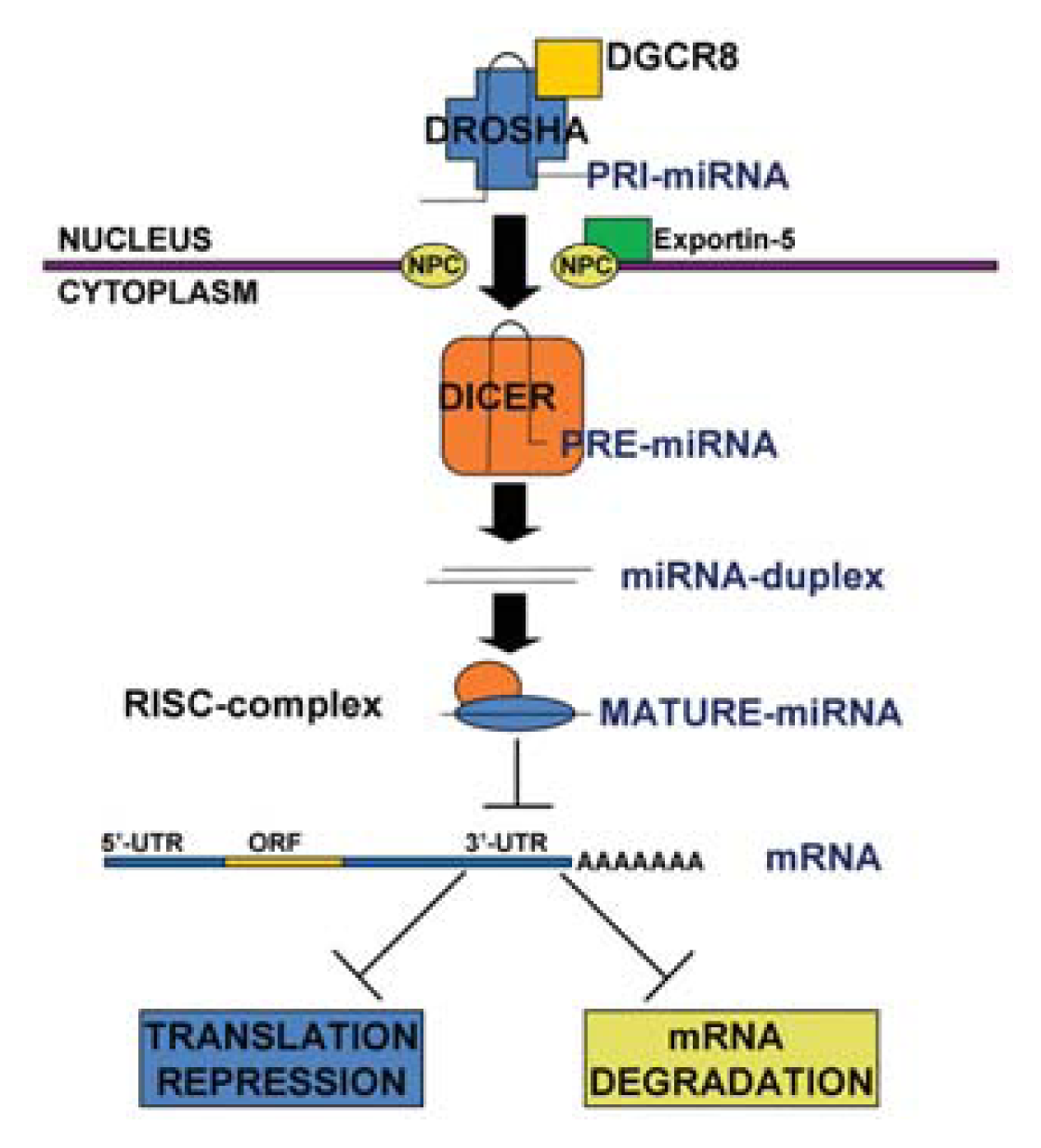
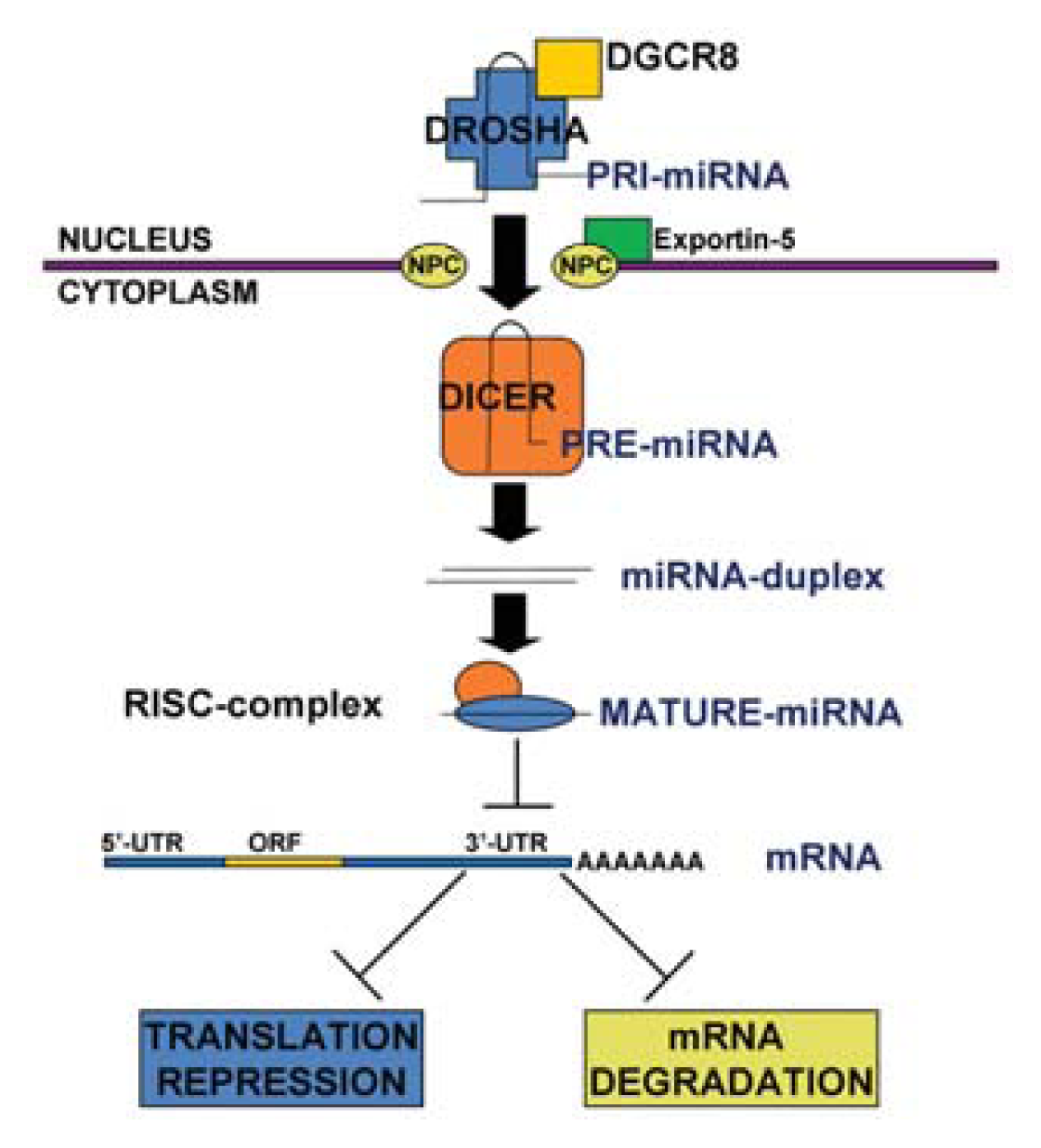
1.1. MicroRNA Biogenesis and Function
1.2. Oxidative Stress and Cardiovascular Diseases
2. miRNAs Modulated by ROS in ECs and VSMCs
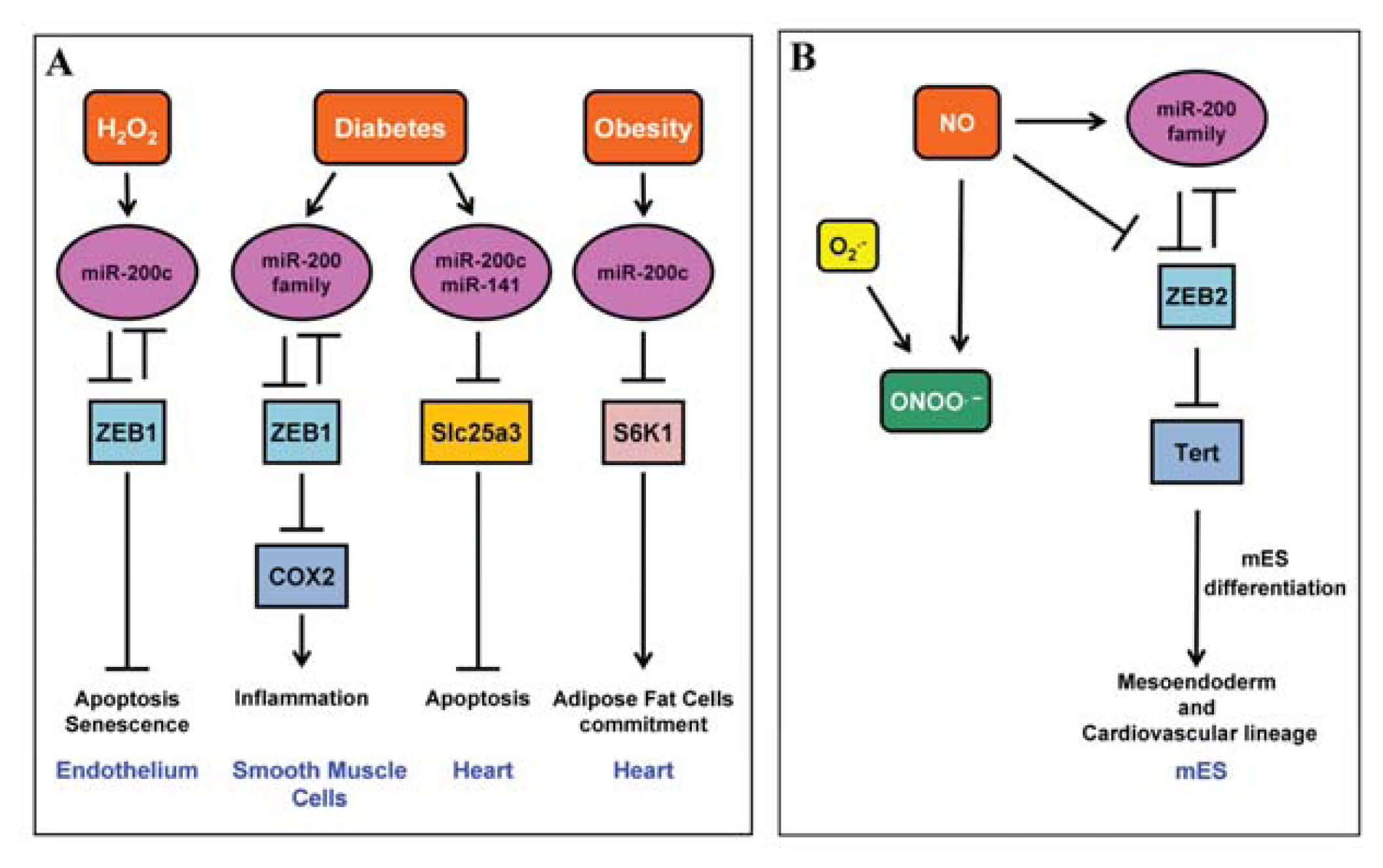
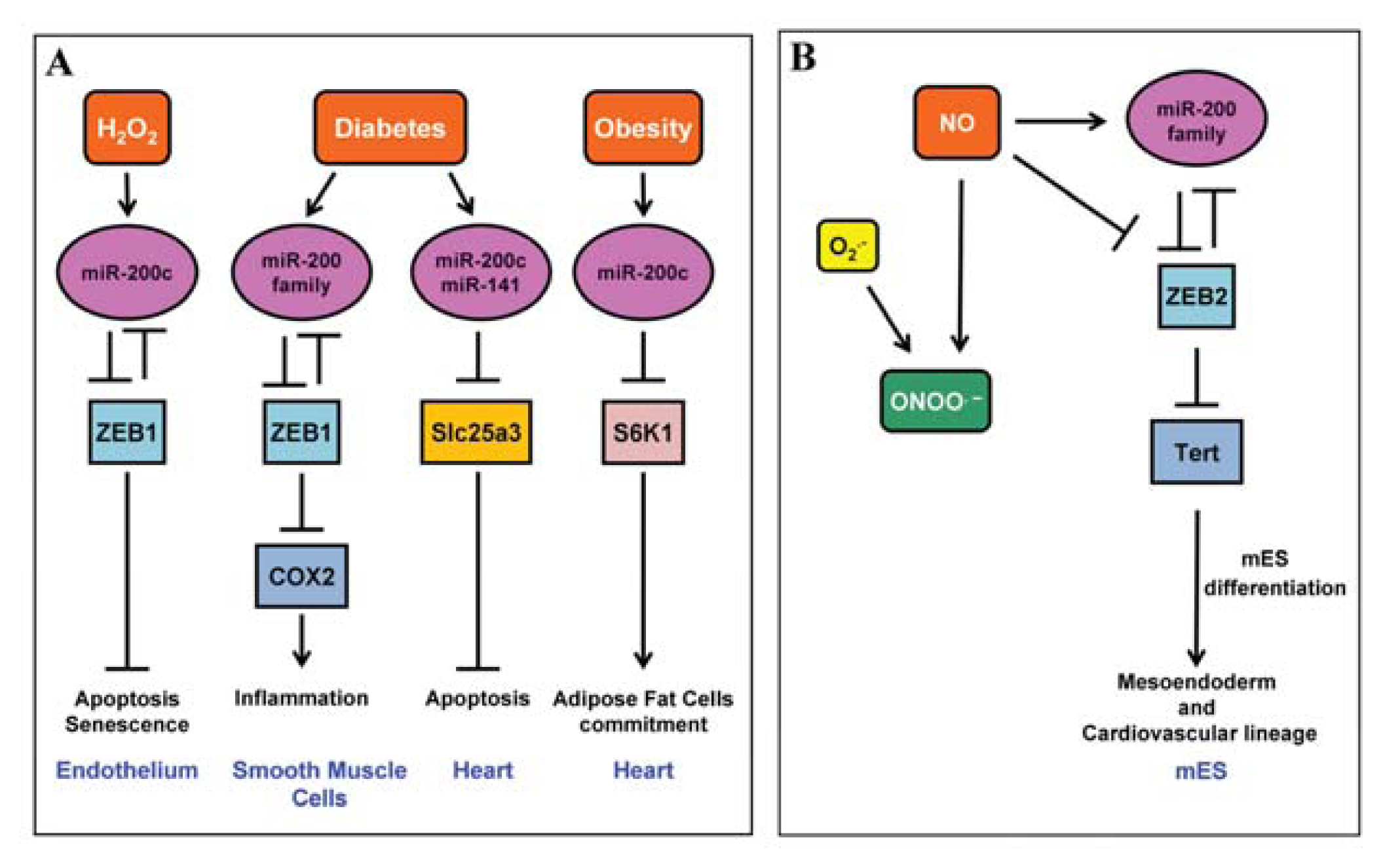
2.1. miR-200 Family
2.2. Sirtuin 1-Targeting miRNAs
2.3. miR-21
3. miRNAs and Mitochondrial Metabolism and Dysfunction
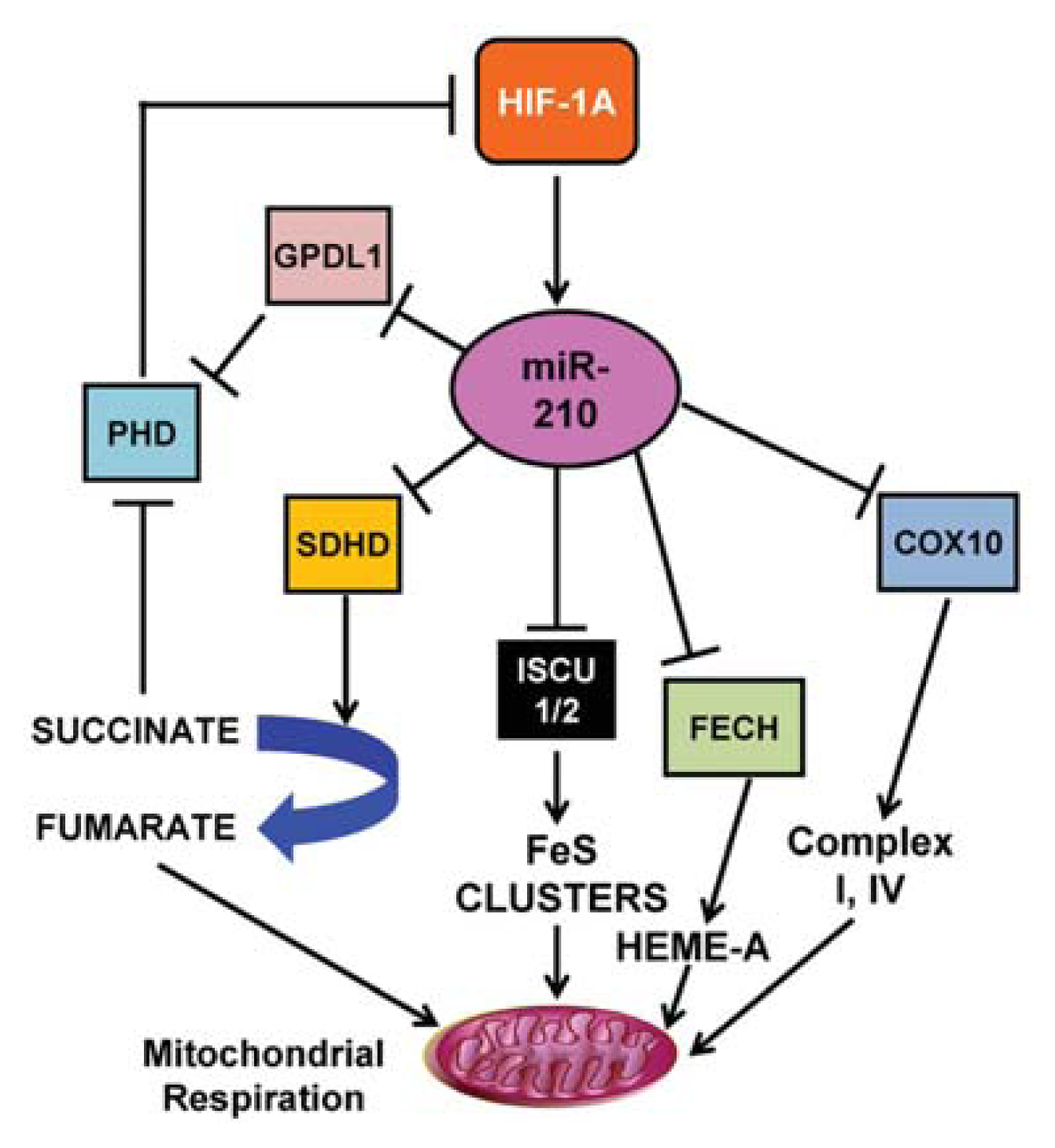
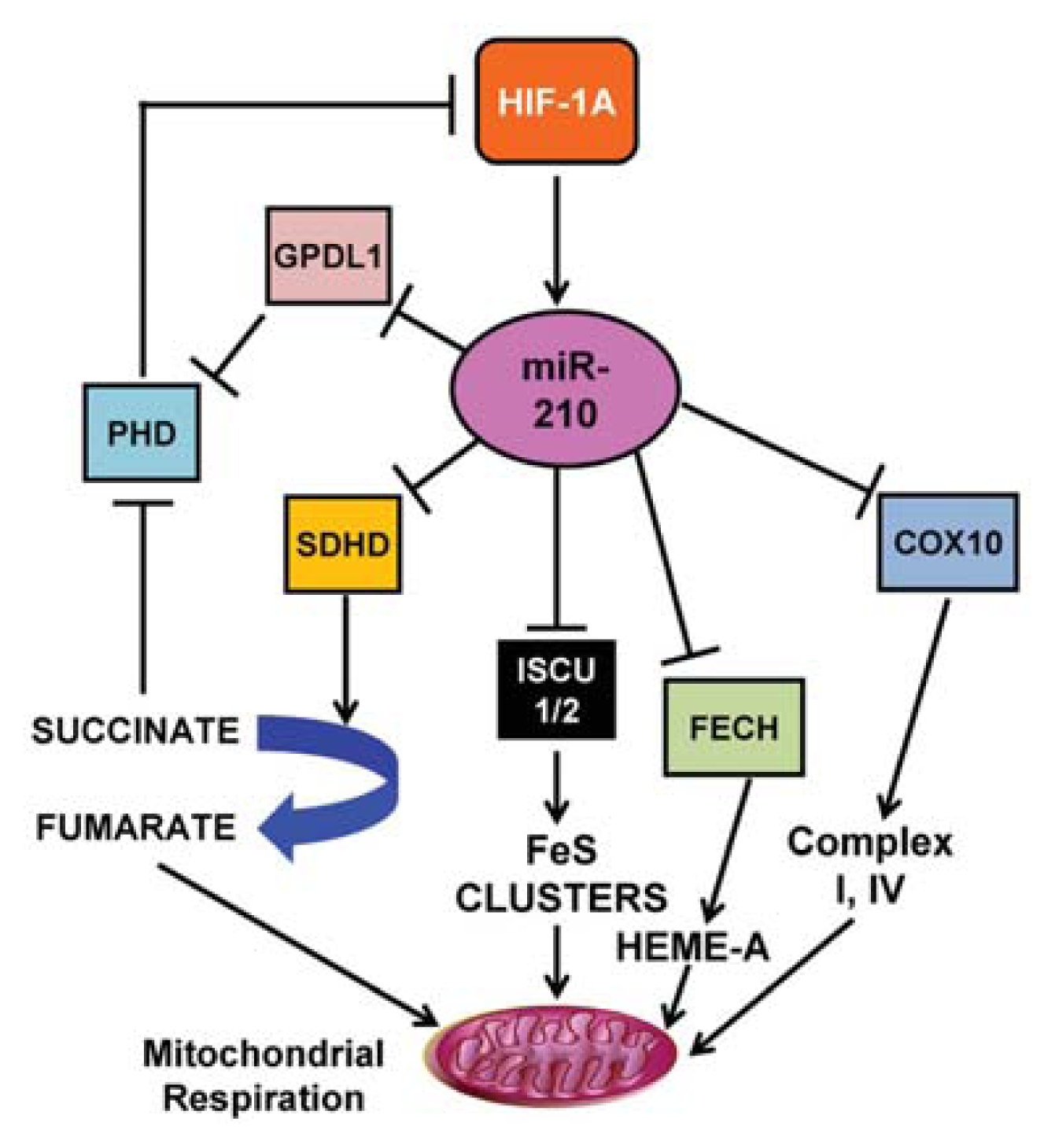
3.1. miR-210
3.2. miR-23a/b
3.3. miRNAs Involved in Mitochondrial ATP Level Regulation
4. miRNAs in Vascular Diseases Associated to Obesity and Diabetes
4.1. miRNAs in Obesity-Associated Systemic Inflammation
4.2. miRNAs and VSMC Dysfunction in Diabetes
4.3. miR-200 Family
5. Challenges and Future Directions
Acknowledgments
Conflicts of interest
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet 2011, 12, 99–110. [Google Scholar]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol 2009, 11, 228–234. [Google Scholar]
- Vasudevan, S. Posttranscriptional upregulation by microRNAs. Wiley Interdiscip. Rev. RNA 2011, 3, 311–330. [Google Scholar]
- Elefant, N.; Altuvia, Y.; Margalit, H. A wide repertoire of miRNA binding sites: Prediction and functional implications. Bioinformatics 2011, 27, 3093–3101. [Google Scholar]
- Fasanaro, P.; Romani, S.; Voellenkle, C.; Maimone, B.; Capogrossi, M.C.; Martelli, F. ROD1 is a seedless target gene of hypoxia-induced miR-210. PLoS One 2012, 7, e44651. [Google Scholar]
- Lal, A.; Navarro, F.; Maher, C.A.; Maliszewski, L.E.; Yan, N.; O’Day, E.; Chowdhury, D.; Dykxhoorn, D.M.; Tsai, P.; Hofmann, O.; et al. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′UTR microRNA recognition elements. Mol. Cell 2009, 35, 610–625. [Google Scholar]
- Kim, G.H.; Ryan, J.J.; Archer, S.L. The role of redox signaling in epigenetics and cardiovascular disease. Antioxid. Redox. Signal 2013, 18, 1920–1936. [Google Scholar]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol 2003, 15, 247–254. [Google Scholar]
- Fridovich, I. The biology of oxygen radicals. Science 1978, 201, 875–880. [Google Scholar]
- Napoli, C.; de Nigris, F.; Palinski, W. Multiple role of reactive oxygen species in the arterial wall. J. Cell. Biochem 2001, 82, 674–682. [Google Scholar]
- Madamanchi, N.R.; Vendrov, A.; Runge, M.S. Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol 2005, 25, 29–38. [Google Scholar]
- Irani, K. Oxidant signaling in vascular cell growth, death, and survival: A review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ. Res 2000, 87, 179–183. [Google Scholar]
- Giorgio, M.; Trinei, M.; Migliaccio, E.; Pelicci, P.G. Hydrogen peroxide: A metabolic by-product or a common mediator of ageing signals? Nat. Rev 2007, 8, 722–728. [Google Scholar]
- Higashi, Y.; Noma, K.; Yoshizumi, M.; Kihara, Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ. J 2009, 73, 411–418. [Google Scholar]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 2004, 109, 227–233. [Google Scholar]
- Fearon, I.M.; Faux, S.P. Oxidative stress and cardiovascular disease: Novel tools give (free) radical insight. J. Mol. Cell. Cardiol 2009, 47, 372–381. [Google Scholar]
- Lassegue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol 2003, 285, R277–R297. [Google Scholar]
- Ellmark, S.H.; Dusting, G.J.; Fui, M.N.; Guzzo-Pernell, N.; Drummond, G.R. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc. Res 2005, 65, 495–504. [Google Scholar]
- Stasi, M.A.; Scioli, M.G.; Arcuri, G.; Mattera, G.G.; Lombardo, K.; Marcellini, M.; Riccioni, T.; de Falco, S.; Pisano, C.; Spagnoli, L.G.; et al. Propionyl-l-carnitine improves postischemic blood flow recovery and arteriogenetic revascularization and reduces endothelial NADPH-oxidase 4-mediated superoxide production. Arterioscler. Thromb. Vasc. Biol 2010, 30, 426–435. [Google Scholar]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med 1999, 340, 115–126. [Google Scholar]
- Yang, Y.M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol 2009, 297, H1829–H1836. [Google Scholar]
- Alp, N.J.; Channon, K.M. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol 2004, 24, 413–420. [Google Scholar]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res 2006, 71, 216–225. [Google Scholar]
- Xu, S.; Zhang, R.; Niu, J.; Cui, D.; Xie, B.; Zhang, B.; Lu, K.; Yu, W.; Wang, X.; Zhang, Q. Oxidative stress mediated-alterations of the MicroRNA expression profile in mouse hippocampal neurons. Int. J. Mol. Sci 2012, 13, 16945–16960. [Google Scholar]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep 2010, 11, 670–677. [Google Scholar]
- Simone, N.L.; Soule, B.P.; Ly, D.; Saleh, A.D.; Savage, J.E.; Degraff, W.; Cook, J.; Harris, C.C.; Gius, D.; Mitchell, J.B. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One 2009, 4, e6377. [Google Scholar]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell. Death Differ 2011, 18, 1628–1639. [Google Scholar]
- Villeneuve, L.M.; Reddy, M.A.; Lanting, L.L.; Wang, M.; Meng, L.; Natarajan, R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc. Natl. Acad. Sci. USA 2008, 105, 9047–9052. [Google Scholar]
- Igosheva, N.; Abramov, A.Y.; Poston, L.; Eckert, J.J.; Fleming, T.P.; Duchen, M.R.; McConnell, J. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS One 2010, 5, e10074. [Google Scholar]
- Suarez, Y.; Fernandez-Hernando, C.; Pober, J.S.; Sessa, W.C. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ. Res 2007, 100, 1164–1173. [Google Scholar]
- Zaccagnini, G.; Martelli, F.; Fasanaro, P.; Magenta, A.; Gaetano, C.; di Carlo, A.; Biglioli, P.; Giorgio, M.; Martin-Padura, I.; Pelicci, P.G.; et al. p66ShcA modulates tissue response to hindlimb ischemia. Circulation 2004, 109, 2917–2923. [Google Scholar]
- Cicchillitti, L.; Di Stefano, V.; Isaia, E.; Crimaldi, L.; Fasanaro, P.; Ambrosino, V.; Antonini, A.; Capogrossi, M.C.; Gaetano, C.; Piaggio, G.; et al. Hypoxia-inducible factor 1-alpha induces miR-210 in normoxic differentiating myoblasts. J. Biol. Chem 2012, 287, 44761–44771. [Google Scholar]
- Chan, S.Y.; Loscalzo, J. MicroRNA-210: A unique and pleiotropic hypoxamir. Cell Cycle 2010, 9, 1072–1083. [Google Scholar]
- Devlin, C.; Greco, S.; Martelli, F.; Ivan, M. miR-210: More than a silent player in hypoxia. IUBMB Life 2011, 63, 94–100. [Google Scholar]
- Liu, M.; Liu, H.; Dudley, S.C., Jr. Reactive oxygen species originating from mitochondria regulate the cardiac sodium channel. Circ. Res. 2010, 107, 967–974. [Google Scholar]
- Ye, H.; Rouault, T.A. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar]
- Tong, W.H.; Rouault, T.A. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab 2006, 3, 199–210. [Google Scholar]
- Qiao, A.; Khechaduri, A.; Kannan Mutharasan, R.; Wu, R.; Nagpal, V.; Ardehali, H. MicroRNA-210 decreases heme levels by targeting ferrochelatase in cardiomyocytes. J. Am. Heart Assoc 2013, 2, e000121. [Google Scholar]
- Puissegur, M.P.; Mazure, N.M.; Bertero, T.; Pradelli, L.; Grosso, S.; Robbe-Sermesant, K.; Maurin, T.; Lebrigand, K.; Cardinaud, B.; Hofman, V.; et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ 2011, 18, 465–478. [Google Scholar]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends Immunol 2004, 25, 4–7. [Google Scholar]
- Chang, C.L.; Au, L.C.; Huang, S.W.; Fai Kwok, C.; Ho, L.T.; Juan, C.C. Insulin up-regulates heme oxygenase-1 expression in 3T3-L1 adipocytes via PI3-kinase- and PKC-dependent pathways and heme oxygenase-1-associated microRNA downregulation. Endocrinology 2011, 152, 384–393. [Google Scholar]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar]
- Hulsmans, M.; Holvoet, P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J. Cell. Mol. Med 2010, 14, 70–78. [Google Scholar]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res 2007, 100, 1512–1521. [Google Scholar]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar]
- Lin, Z.; Murtaza, I.; Wang, K.; Jiao, J.; Gao, J.; Li, P.F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 12103–12108. [Google Scholar]
- Aubert, G.; Vega, R.B.; Kelly, D.P. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim. Biophys. Acta 2013, 1833, 840–847. [Google Scholar]
- Shen, X.; Zheng, S.; Thongboonkerd, V.; Xu, M.; Pierce, W.M., Jr; Klein, J.B.; Epstein, P.N. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E896–E905. [Google Scholar]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar]
- Tan, G.; Shi, Y.; Wu, Z.H. MicroRNA-22 promotes cell survival upon UV radiation by repressing PTEN. Biochem. Biophys. Res. Commun 2012, 417, 546–551. [Google Scholar]
- Schickel, R.; Park, S.M.; Murmann, A.E.; Peter, M.E. miR-200c regulates induction of apoptosis through CD95 by targeting FAP-1. Mol. Cell 2010, 38, 908–915. [Google Scholar]
- Li, G.; Luna, C.; Qiu, J.; Epstein, D.L.; Gonzalez, P. Alterations in microRNA expression in stress-induced cellular senescence. Mech. Ageing Dev 2009, 130, 731–741. [Google Scholar]
- Wang, Z.; Liu, Y.; Han, N.; Chen, X.; Yu, W.; Zhang, W.; Zou, F. Profiles of oxidative stress-related microRNA and mRNA expression in auditory cells. Brain Res 2010, 1346, 14–25. [Google Scholar]
- Rosati, J.; Spallotta, F.; Nanni, S.; Grasselli, A.; Antonini, A.; Vincenti, S.; Presutti, C.; Colussi, C.; D’Angelo, C.; Biroccio, A.; et al. Smad-interacting protein-1 and microRNA 200 family define a nitric oxide-dependent molecular circuitry involved in embryonic stem cell mesendoderm differentiation. Arterioscler. Thromb. Vasc. Biol 2011, 31, 898–907. [Google Scholar]
- Mateescu, B.; Batista, L.; Cardon, M.; Gruosso, T.; de Feraudy, Y.; Mariani, O.; Nicolas, A.; Meyniel, J.P.; Cottu, P.; Sastre-Garau, X.; et al. miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med 2011, 17, 1627–1635. [Google Scholar]
- Dolado, I.; Swat, A.; Ajenjo, N.; de Vita, G.; Cuadrado, A.; Nebreda, A.R. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell 2007, 11, 191–205. [Google Scholar]
- Hui, L.; Bakiri, L.; Mairhorfer, A.; Schweifer, N.; Haslinger, C.; Kenner, L.; Komnenovic, V.; Scheuch, H.; Beug, H.; Wagner, E.F. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet 2007, 39, 741–749. [Google Scholar]
- Kennedy, N.J.; Cellurale, C.; Davis, R.J. A radical role for p38 MAPK in tumor initiation. Cancer Cell 2007, 11, 101–103. [Google Scholar]
- Naidu, S.; Vijayan, V.; Santoso, S.; Kietzmann, T.; Immenschuh, S. Inhibition and genetic deficiency of p38 MAPK up-regulates heme oxygenase-1 gene expression via Nrf2. J. Immunol 2009, 182, 7048–7057. [Google Scholar]
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003. [Google Scholar]
- Schwer, B.; Verdin, E. Conserved metabolic regulatory functions of sirtuins. Cell Metab 2008, 7, 104–112. [Google Scholar]
- Chen, Z.; Shentu, T.P.; Wen, L.; Johnson, D.A.; Shyy, J.Y. Regulation of SIRT1 by oxidative stress-responsive miRNAs and a systematic approach to identify its role in the endothelium. Antioxid. Redox. Signal. 2013. [Google Scholar] [CrossRef]
- Potente, M.; Dimmeler, S. Emerging roles of SIRT1 in vascular endothelial homeostasis. Cell Cycle 2008, 7, 2117–2122. [Google Scholar]
- Rane, S.; He, M.; Sayed, D.; Vashistha, H.; Malhotra, A.; Sadoshima, J.; Vatner, D.E.; Vatner, S.F.; Abdellatif, M. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ. Res 2009, 104, 879–886. [Google Scholar]
- Xu, Q.; Seeger, F.H.; Castillo, J.; Iekushi, K.; Boon, R.A.; Farcas, R.; Manavski, Y.; Li, Y.G.; Assmus, B.; Zeiher, A.M.; et al. Micro-RNA-34a contributes to the impaired function of bone marrow-derived mononuclear cells from patients with cardiovascular disease. J. Am. Coll. Cardiol 2012, 59, 2107–2117. [Google Scholar]
- Eades, G.; Yao, Y.; Yang, M.; Zhang, Y.; Chumsri, S.; Zhou, Q. miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J. Biol. Chem 2011, 286, 25992–26002. [Google Scholar]
- Lin, Y.; Liu, X.; Cheng, Y.; Yang, J.; Huo, Y.; Zhang, C. Involvement of MicroRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J. Biol. Chem 2009, 284, 7903–7913. [Google Scholar]
- Yang, H.S.; Knies, J.L.; Stark, C.; Colburn, N.H. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene 2003, 22, 3712–3720. [Google Scholar]
- Weber, M.; Baker, M.B.; Moore, J.P.; Searles, C.D. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem. Biophys. Res. Commun 2010, 393, 643–648. [Google Scholar]
- Fleissner, F.; Jazbutyte, V.; Fiedler, J.; Gupta, S.K.; Yin, X.; Xu, Q.; Galuppo, P.; Kneitz, S.; Mayr, M.; Ertl, G.; et al. Short communication: Asymmetric dimethylarginine impairs angiogenic progenitor cell function in patients with coronary artery disease through a microRNA-21-dependent mechanism. Circ. Res 2010, 107, 138–143. [Google Scholar]
- Semenza, G.L. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J 2007, 405, 1–9. [Google Scholar]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med 2011, 365, 537–547. [Google Scholar]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol 2011, 300, C385–C393. [Google Scholar]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H2181–H2190. [Google Scholar]
- Perrelli, M.G.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol 2011, 3, 186–200. [Google Scholar]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem 2008, 283, 10892–10903. [Google Scholar]
- Prabhakar, N.R.; Semenza, G.L. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol. Rev 2012, 92, 967–1003. [Google Scholar]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006, 3, 177–185. [Google Scholar]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab 2009, 10, 273–284. [Google Scholar]
- Fasanaro, P.; Greco, S.; Lorenzi, M.; Pescatori, M.; Brioschi, M.; Kulshreshtha, R.; Banfi, C.; Stubbs, A.; Calin, G.A.; Ivan, M.; et al. An integrated approach for experimental target identification of hypoxia-induced miR-210. J. Biol. Chem 2009, 284, 35134–35143. [Google Scholar]
- Yoshioka, Y.; Kosaka, N.; Ochiya, T.; Kato, T. Micromanaging Iron Homeostasis: Hypoxia-inducible micro-RNA-210 suppresses iron homeostasis-related proteins. J. Biol. Chem 2012, 287, 34110–34119. [Google Scholar]
- Schmucker, S.; Puccio, H. Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum. Mol. Genet 2010, 19, R103–R110. [Google Scholar]
- Diaz, F.; Fukui, H.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol. Cell Biol 2006, 26, 4872–4881. [Google Scholar]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar]
- Giannakakis, A.; Sandaltzopoulos, R.; Greshock, J.; Liang, S.; Huang, J.; Hasegawa, K.; Li, C.; O’Brien-Jenkins, A.; Katsaros, D.; Weber, B.L.; et al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther 2008, 7, 255–264. [Google Scholar]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landazuri, M.O.; Enriquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab 2012, 16, 378–386. [Google Scholar]
- Kelly, T.J.; Souza, A.L.; Clish, C.B.; Puigserver, P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol. Cell Biol 2011, 31, 2696–2706. [Google Scholar]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.L.; et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS One 2010, 5, e10345. [Google Scholar]
- Mutharasan, R.K.; Nagpal, V.; Ichikawa, Y.; Ardehali, H. microRNA-210 is upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects. Am. J. Physiol. Heart Circ. Physiol 2011, 301, H1519–H1530. [Google Scholar]
- Fasanaro, P.; D’Alessandra, Y.; di Stefano, V.; Melchionna, R.; Romani, S.; Pompilio, G.; Capogrossi, M.C.; Martelli, F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J. Biol. Chem 2008, 283, 15878–15883. [Google Scholar]
- Kim, J.H.; Park, S.G.; Song, S.Y.; Kim, J.K.; Sung, J.H. Reactive oxygen species-responsive miR-210 regulates proliferation and migration of adipose-derived stem cells via PTPN2. Cell Death Dis 2013, 4, e588. [Google Scholar]
- Faraonio, R.; Salerno, P.; Passaro, F.; Sedia, C.; Iaccio, A.; Bellelli, R.; Nappi, T.C.; Comegna, M.; Romano, S.; Salvatore, G.; et al. A set of miRNAs participates in the cellular senescence program in human diploid fibroblasts. Cell Death Differ 2012, 19, 713–721. [Google Scholar]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med 2006, 50, 567–575. [Google Scholar]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. (Berl. ) 2010, 88, 993–1001. [Google Scholar]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; de Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar]
- Lin, H.; Qian, J.; Castillo, A.C.; Long, B.; Keyes, K.T.; Chen, G.; Ye, Y. Effect of miR-23 on oxidant-induced injury in human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci 2011, 52, 6308–6314. [Google Scholar]
- Wilson, F.H.; Hariri, A.; Farhi, A.; Zhao, H.; Petersen, K.F.; Toka, H.R.; Nelson-Williams, C.; Raja, K.M.; Kashgarian, M.; Shulman, G.I.; et al. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science 2004, 306, 1190–1194. [Google Scholar]
- Nishi, H.; Ono, K.; Iwanaga, Y.; Horie, T.; Nagao, K.; Takemura, G.; Kinoshita, M.; Kuwabara, Y.; Mori, R.T.; Hasegawa, K.; et al. MicroRNA-15b modulates cellular ATP levels and degenerates mitochondria via Arl2 in neonatal rat cardiac myocytes. J. Biol. Chem 2010, 285, 4920–4930. [Google Scholar]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar]
- Hullinger, T.G.; Montgomery, R.L.; Seto, A.G.; Dickinson, B.A.; Semus, H.M.; Lynch, J.M.; Dalby, C.M.; Robinson, K.; Stack, C.; Latimer, P.A.; et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res 2010, 110, 71–81. [Google Scholar]
- Spinetti, G.; Fortunato, O.; Caporali, A.; Shantikumar, S.; Marchetti, M.; Meloni, M.; Descamps, B.; Floris, I.; Sangalli, E.; Vono, R.; et al. MicroRNA-15a and microRNA-16 impair human circulating proangiogenic cell functions and are increased in the proangiogenic cells and serum of patients with critical limb ischemia. Circ. Res 2013, 112, 335–346. [Google Scholar]
- Keaney, J.F., Jr; Larson, M.G.; Vasan, R.S.; Wilson, P.W.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J. Obesity and systemic oxidative stress: Clinical correlates of oxidative stress in the Framingham Study. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar]
- Whiting, P.H.; Kalansooriya, A.; Holbrook, I.; Haddad, F.; Jennings, P.E. The relationship between chronic glycaemic control and oxidative stress in type 2 diabetes mellitus. Br. J. Biomed. Sci 2008, 65, 71–74. [Google Scholar]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res 2005, 96, 939–949. [Google Scholar]
- Mangiapane, H. Cardiovascular disease and diabetes. Adv. Exp. Med. Biol 2012, 771, 219–228. [Google Scholar]
- Bruhn-Olszewska, B.; Korzon-Burakowska, A.; Gabig-Ciminska, M.; Olszewski, P.; Wegrzyn, A.; Jakobkiewicz-Banecka, J. Molecular factors involved in the development of diabetic foot syndrome. Acta Biochim. Pol 2012, 59, 507–513. [Google Scholar]
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol 2006, 212, 167–178. [Google Scholar]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res 2010, 107, 1058–1070. [Google Scholar]
- Ortega, F.J.; Moreno-Navarrete, J.M.; Pardo, G.; Sabater, M.; Hummel, M.; Ferrer, A.; Rodriguez-Hermosa, J.I.; Ruiz, B.; Ricart, W.; Peral, B.; et al. MiRNA expression profile of human subcutaneous adipose and during adipocyte differentiation. PLoS One 2010, 5, e9022. [Google Scholar]
- Liu, X.; Cheng, Y.; Yang, J.; Xu, L.; Zhang, C. Cell-specific effects of miR-221/222 in vessels: Molecular mechanism and therapeutic application. J. Mol. Cell. Cardiol 2012, 52, 245–255. [Google Scholar]
- Poliseno, L.; Tuccoli, A.; Mariani, L.; Evangelista, M.; Citti, L.; Woods, K.; Mercatanti, A.; Hammond, S.; Rainaldi, G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood 2006, 108, 3068–3071. [Google Scholar]
- Lin, Q.; Gao, Z.; Alarcon, R.M.; Ye, J.; Yun, Z. A role of miR-27 in the regulation of adipogenesis. FEBS J 2009, 276, 2348–2358. [Google Scholar]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol 2008, 28, 812–819. [Google Scholar]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar]
- Reddy, M.A.; Jin, W.; Villeneuve, L.; Wang, M.; Lanting, L.; Todorov, I.; Kato, M.; Natarajan, R. Pro-inflammatory role of microrna-200 in vascular smooth muscle cells from diabetic mice. Arterioscler. Thromb. Vasc. Biol 2012, 32, 721–729. [Google Scholar]
- Reddy, M.A.; Natarajan, R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc. Res 2011, 90, 421–429. [Google Scholar]
- Villeneuve, L.M.; Kato, M.; Reddy, M.A.; Wang, M.; Lanting, L.; Natarajan, R. Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes 2010, 59, 2904–2915. [Google Scholar]
- Manca, S.; Magrelli, A.; Cialfi, S.; Lefort, K.; Ambra, R.; Alimandi, M.; Biolcati, G.; Uccelletti, D.; Palleschi, C.; Screpanti, I.; et al. Oxidative stress activation of miR-125b is part of the molecular switch for Hailey-Hailey disease manifestation. Exp. Dermatol 2011, 20, 932–937. [Google Scholar]
- Pulakat, L.; Aroor, A.R.; Gul, R.; Sowers, J.R. Cardiac insulin resistance and microRNA modulators. Exp. Diabetes Res 2012, 2012, 654904. [Google Scholar]
- Kirby, D.M.; Thorburn, D.R. Approaches to finding the molecular basis of mitochondrial oxidative phosphorylation disorders. Twin Res. Hum. Genet 2008, 11, 395–411. [Google Scholar]
- Kiryu-Seo, S.; Gamo, K.; Tachibana, T.; Tanaka, K.; Kiyama, H. Unique anti-apoptotic activity of EAAC1 in injured motor neurons. EMBO J 2006, 25, 3411–3421. [Google Scholar]
- Flarsheim, C.E.; Grupp, I.L.; Matlib, M.A. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am. J. Physiol 1996, 271, H192–H202. [Google Scholar]
- Baseler, W.A.; Thapa, D.; Jagannathan, R.; Dabkowski, E.R.; Croston, T.L.; Hollander, J.M. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am. J. Physiol 2012, 303, C1244–C1251. [Google Scholar]
- Baseler, W.A.; Dabkowski, E.R.; Williamson, C.L.; Croston, T.L.; Thapa, D.; Powell, M.J.; Razunguzwa, T.T.; Hollander, J.M. Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: Contribution of protein import dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol 2011, 300, R186–R200. [Google Scholar]
- Alcala, S.; Klee, M.; Fernandez, J.; Fleischer, A.; Pimentel-Muinos, F.X. A high-throughput screening for mammalian cell death effectors identifies the mitochondrial phosphate carrier as a regulator of cytochrome c release. Oncogene 2008, 27, 44–54. [Google Scholar]
- Ferraro, F.; Lymperi, S.; Mendez-Ferrer, S.; Saez, B.; Spencer, J.A.; Yeap, B.Y.; Masselli, E.; Graiani, G.; Prezioso, L.; Rizzini, E.L.; et al. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci. Transl. Med 2012, 3, 104r, a101.. [Google Scholar]
- Saito, H.; Yamamoto, Y.; Yamamoto, H. Diabetes alters subsets of endothelial progenitor cells that reside in blood, bone marrow, and spleen. Am. J. Physiol 2012, 302, C892–C901. [Google Scholar]
- Fadini, G.P.; Losordo, D.; Dimmeler, S. Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ. Res 2012, 110, 624–637. [Google Scholar]
- Hazarika, S.; Farber, C.R.; Dokun, A.O.; Pitsillides, A.N.; Wang, T.; Lye, R.J.; Annex, B.H. MicroRNA-93 controls perfusion recovery after hindlimb ischemia by modulating expression of multiple genes in the cell cycle pathway. Circulation 2013, 127, 1818–1828. [Google Scholar]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med 2013, 368, 1685–1694. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| ROS source/pathology | miRNAs upregulated | Tissue/organ | Source | Target | Functions | References |
|---|---|---|---|---|---|---|
| H2O2 | miR-200c miR-141 miR-200a miR-200b miR-429 | endothelium, myoblasts | Human | ZEB1 | apoptosis, senescence | [22] |
| H2O2 | miR-200c miR-141 miR-200a miR-200b miR-429 | ovarian adenocarcinomas | Human | p38α | ROS accumulation; improved response to chemotherapy | [25] |
| H2O2 | miR-200c | primary hippocampal neurons | Mouse | Unknown | Unknown | [26] |
| chronic H2O2 treatment | miR-200c | trabecular meshwork cells | Human | Unknown | senescence | [27] |
| t-BHP | miR-200c miR-141 | auditory cells | Mouse | Unknown | Unknown | [28] |
| Obesity | miR-200c miR-141 | Heart | Rat | S6K1 | compensatory/adaptive mechanisms | [29] |
| Diabetes | miR-200c miR-141 | Heart | Mouse | Slc25a3 | dysregulation ATP production; cell death | [30] |
| Diabetes | miR-200c miR-200b miR-429 | VSMCs | Mouse | ZEB1 | inflammation | [31] |
| NO | miR-200c miR-200a miR-200b miR-429 | mES | Mouse | ZEB2 | mesendoderm and cardiovascular differentiation | [32] |
| Hypoxia/ROS | miR-210 | ASCs | Human | PTPN2 | proliferation, migration | [33] |
| Hypoxia | miR-210 | ECs, breast and colon cancer cells | Human | ISCU1/2 | mitochondrial respiration | [34–36] |
| Hypoxia | miR-210 | breast cancer cells | Human | TfR1 | mitochondrial respiration; proliferation | [37] |
| Hypoxia | miR-210 | H9c2 Cardiomyocytes | Mouse | FECH | heme biosynthesis; Iron homeostasis | [38] |
| Hypoxia | miR-210 | colon, breast, esophageal cancer cells | Human | COX10 | mitochondrial respiration; ROS production | [39] |
| Hypoxia | miR-210 | lung cancer cells | Human | SDHD | mitochondrial respiration; proliferation | [40] |
| Hypoxia | miR-210 | ovarian cancer cells | Human | NDUFA4 | mitochondrial respiration | [41] |
| Diabetes | miR-125 | VSMCs | Mouse | Suv39h1 | inflammation | [42] |
| Obesity | miR-27 | Adipose tissue | Mouse | PPARγ, C/EBPα | inflammation | [43] |
| H2O2 | miR-21 | VSMCs | Rat | PDCD4 | apoptosis protection | [44] |
| Coronary artery disease | miR-21 | APCs | Human | SOD-2 SPRY-1 | ROS production; APC migratory defects | [45] |
| Atherosclerosis | miR-217 | Atherosclerotic plaques | Human | SIRT-1 | endothelial dysfunction | [46] |
| Myocardial infarction | miR-34 | BMCs | Human | SIRT-1 | apoptosis | [47] |
| Mitochondrial dysfunction | miR-23a/b | B lymphoma, prostate cancer cells | Human | Mitochondrial GLS | ROS production | [48] |
| Mitochondrial dysfunction | miR-15 family, miR-424 | Cardiomyocytes | Rat | Arl2 | ATP reduction | [49] |
| downregulated: | ||||||
| Obesity | miR-155 miR-183 miR-872 | Adipose tissue | Rat | HO-1 | inflammation, oxidative damage, apoptosis | [50] |
| Hypoxic preconditioning | miR-199a | Cardiomyocytes | Rat | SIRT-1 | apoptosis protection | [51] |
| H2O2 | miR-23a/b | Retinal pigment epithelial cells | Human | Fas | apoptosis | [52] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Magenta, A.; Greco, S.; Gaetano, C.; Martelli, F. Oxidative Stress and MicroRNAs in Vascular Diseases. Int. J. Mol. Sci. 2013, 14, 17319-17346. https://doi.org/10.3390/ijms140917319
Magenta A, Greco S, Gaetano C, Martelli F. Oxidative Stress and MicroRNAs in Vascular Diseases. International Journal of Molecular Sciences. 2013; 14(9):17319-17346. https://doi.org/10.3390/ijms140917319
Chicago/Turabian StyleMagenta, Alessandra, Simona Greco, Carlo Gaetano, and Fabio Martelli. 2013. "Oxidative Stress and MicroRNAs in Vascular Diseases" International Journal of Molecular Sciences 14, no. 9: 17319-17346. https://doi.org/10.3390/ijms140917319