Liberation of GPI-Anchored Prion from Phospholipids Accelerates Amyloidogenic Conversion

Abstract
:
{kind=link}
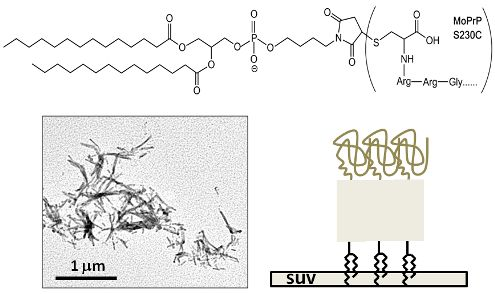{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
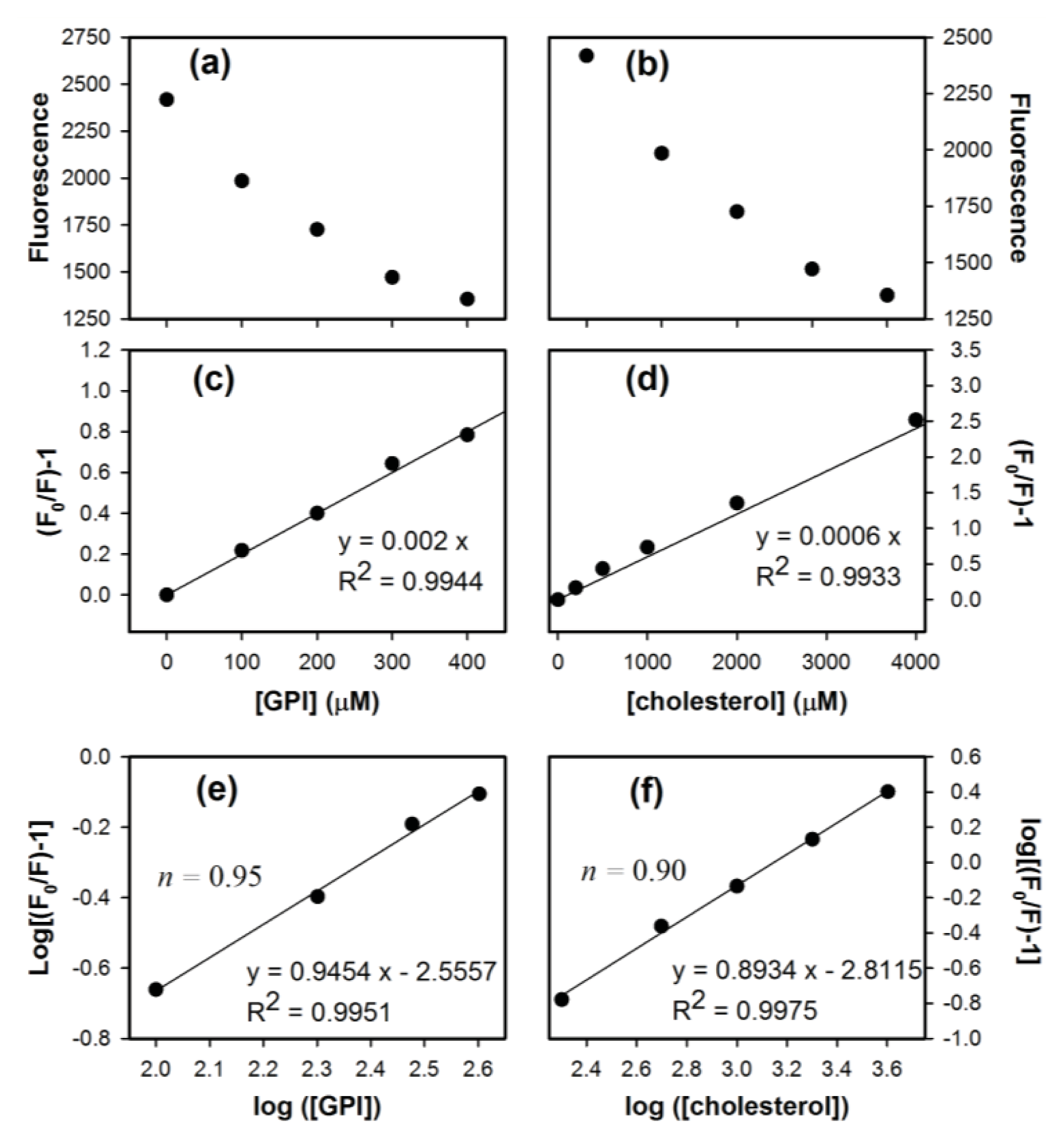
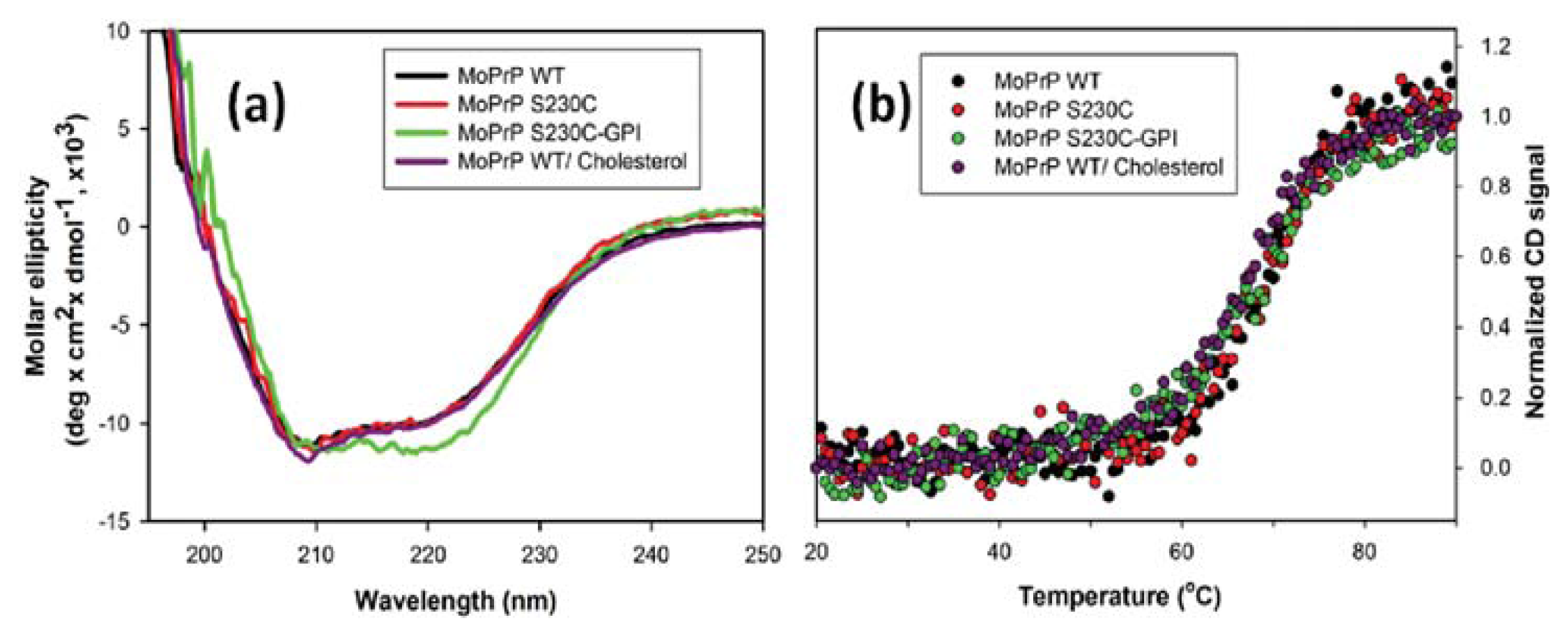
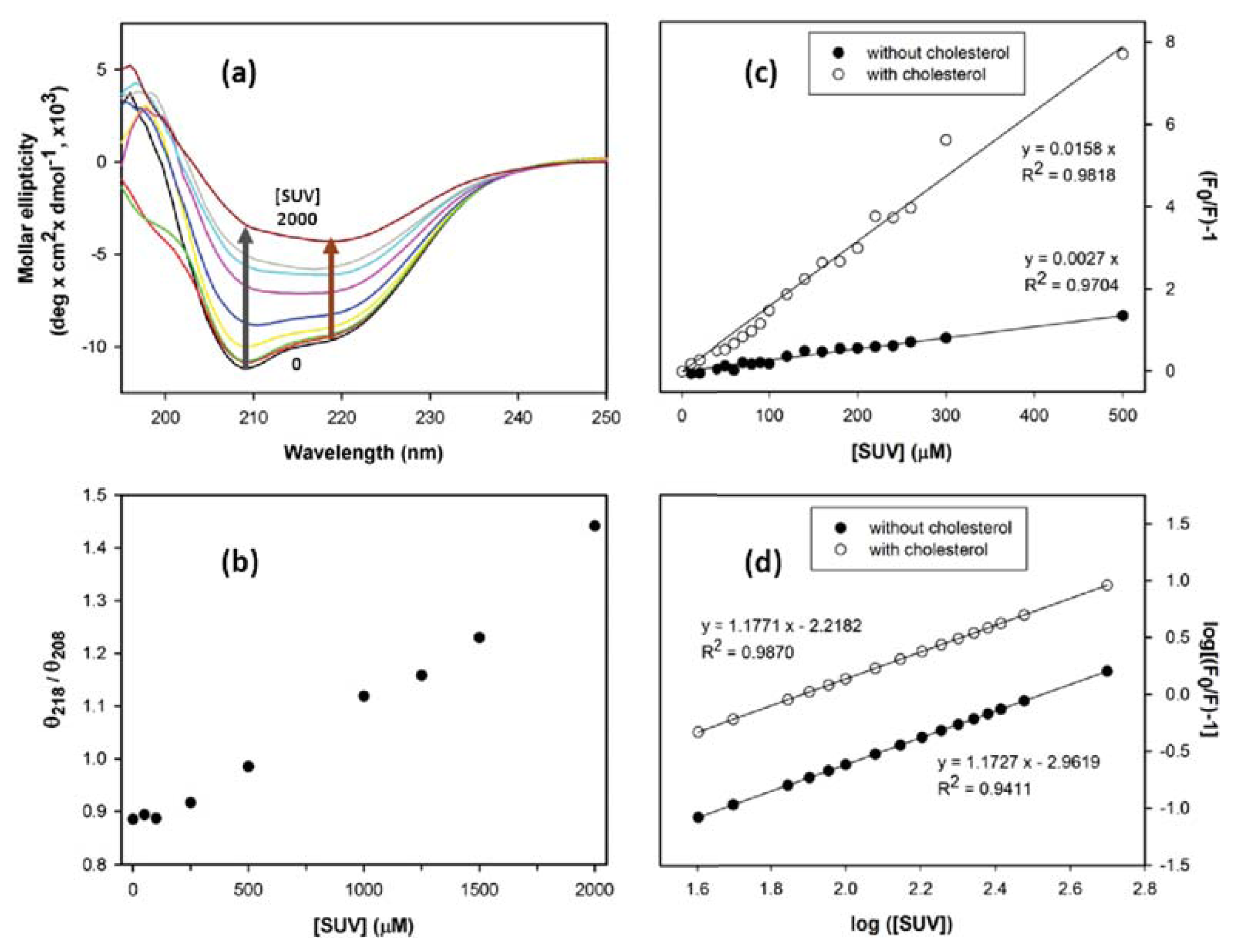
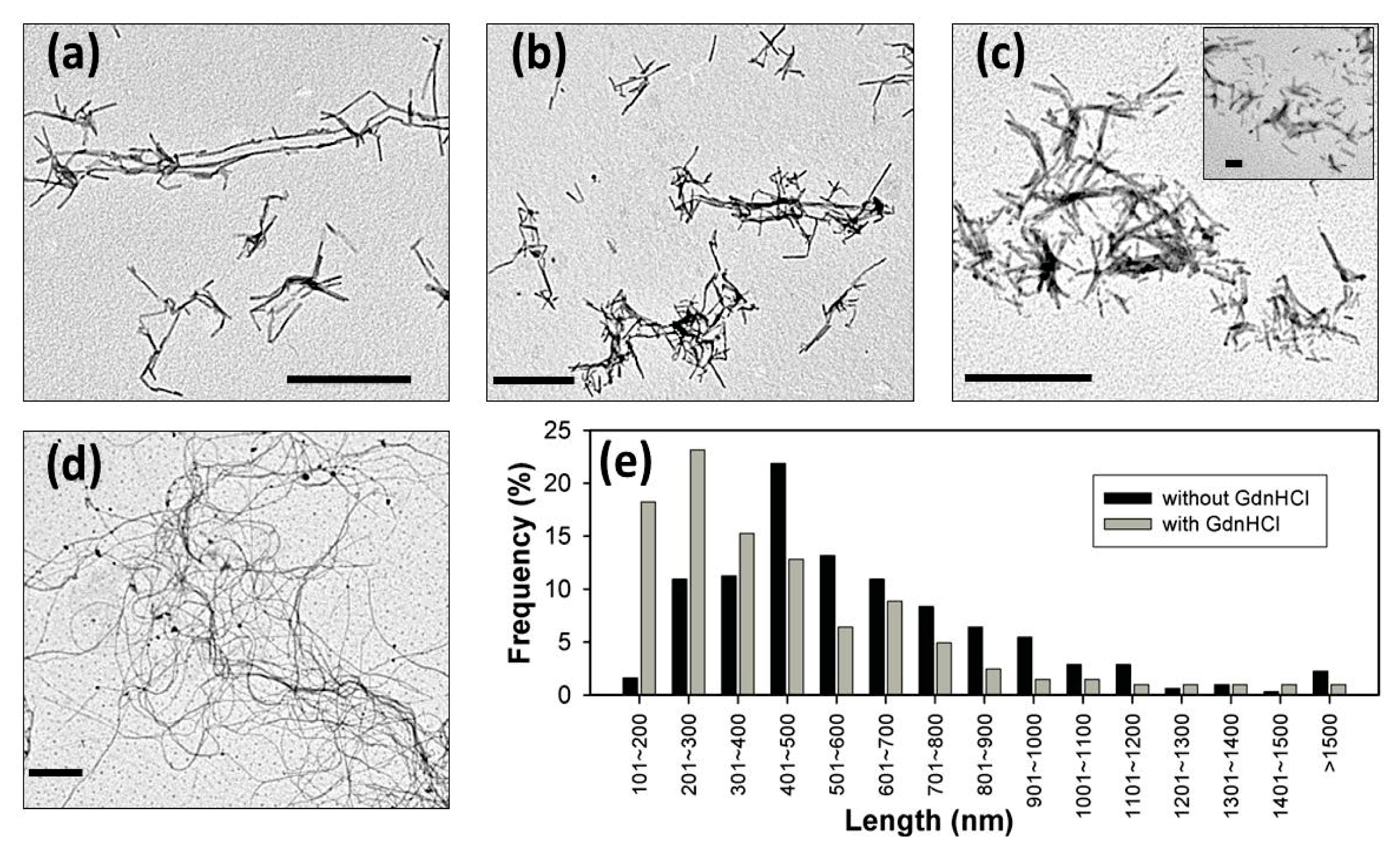
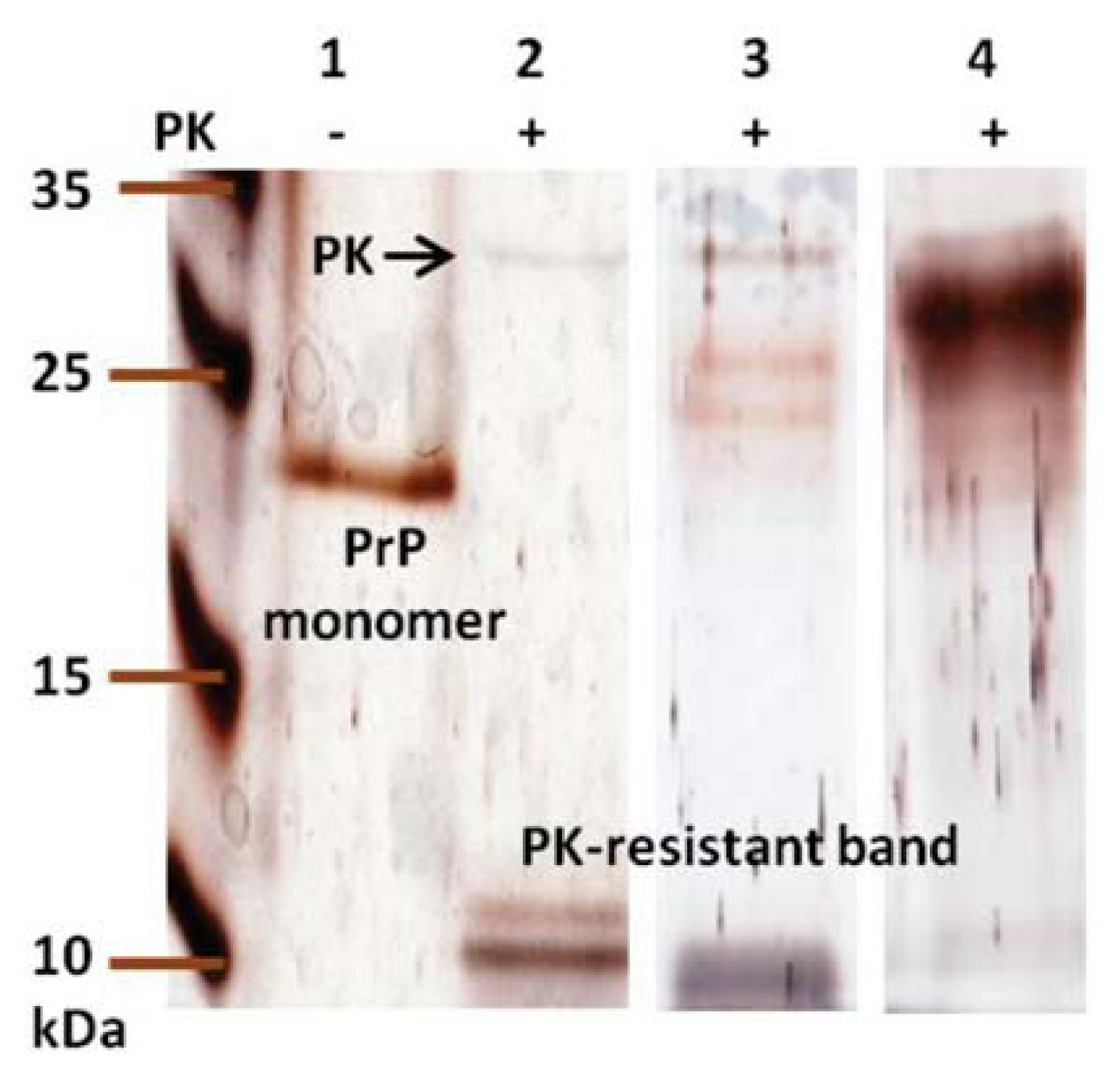
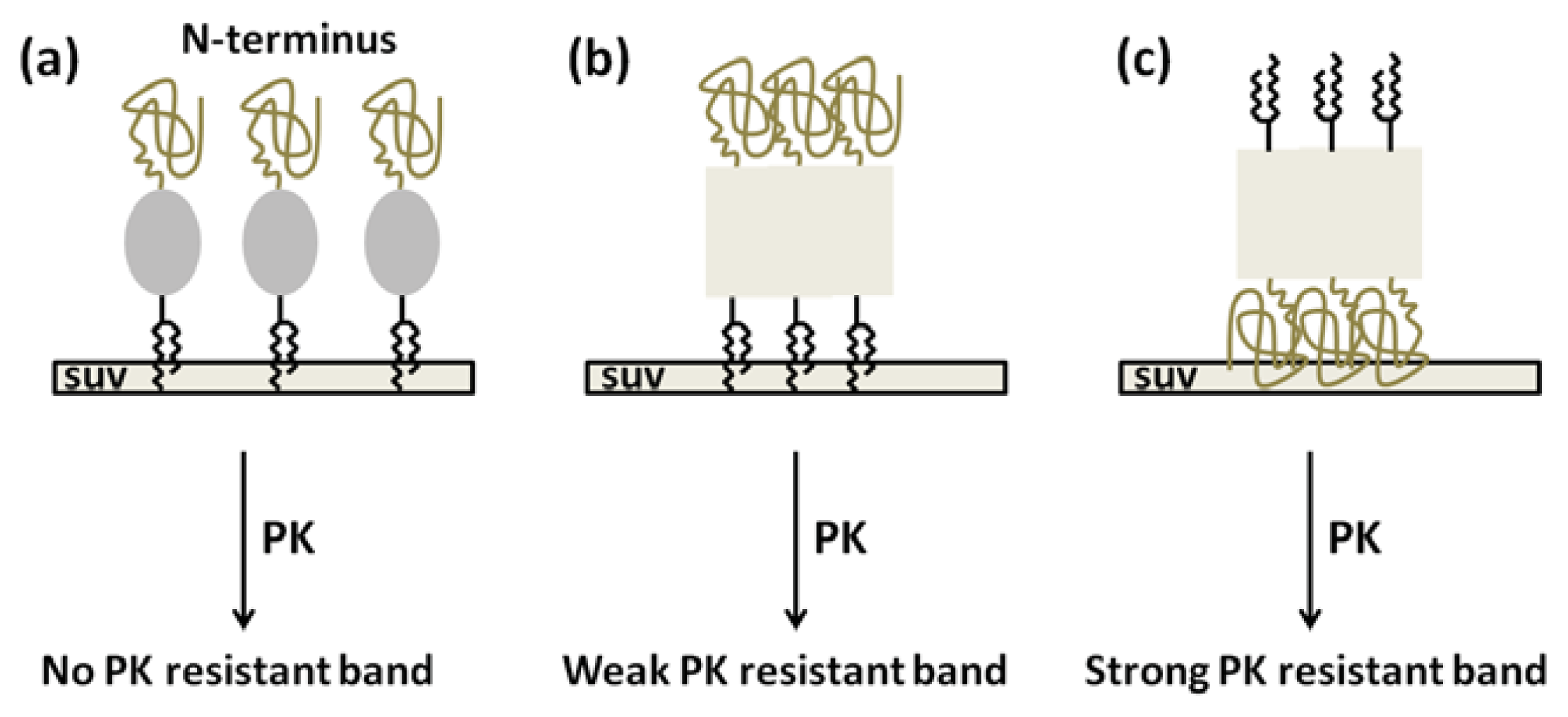
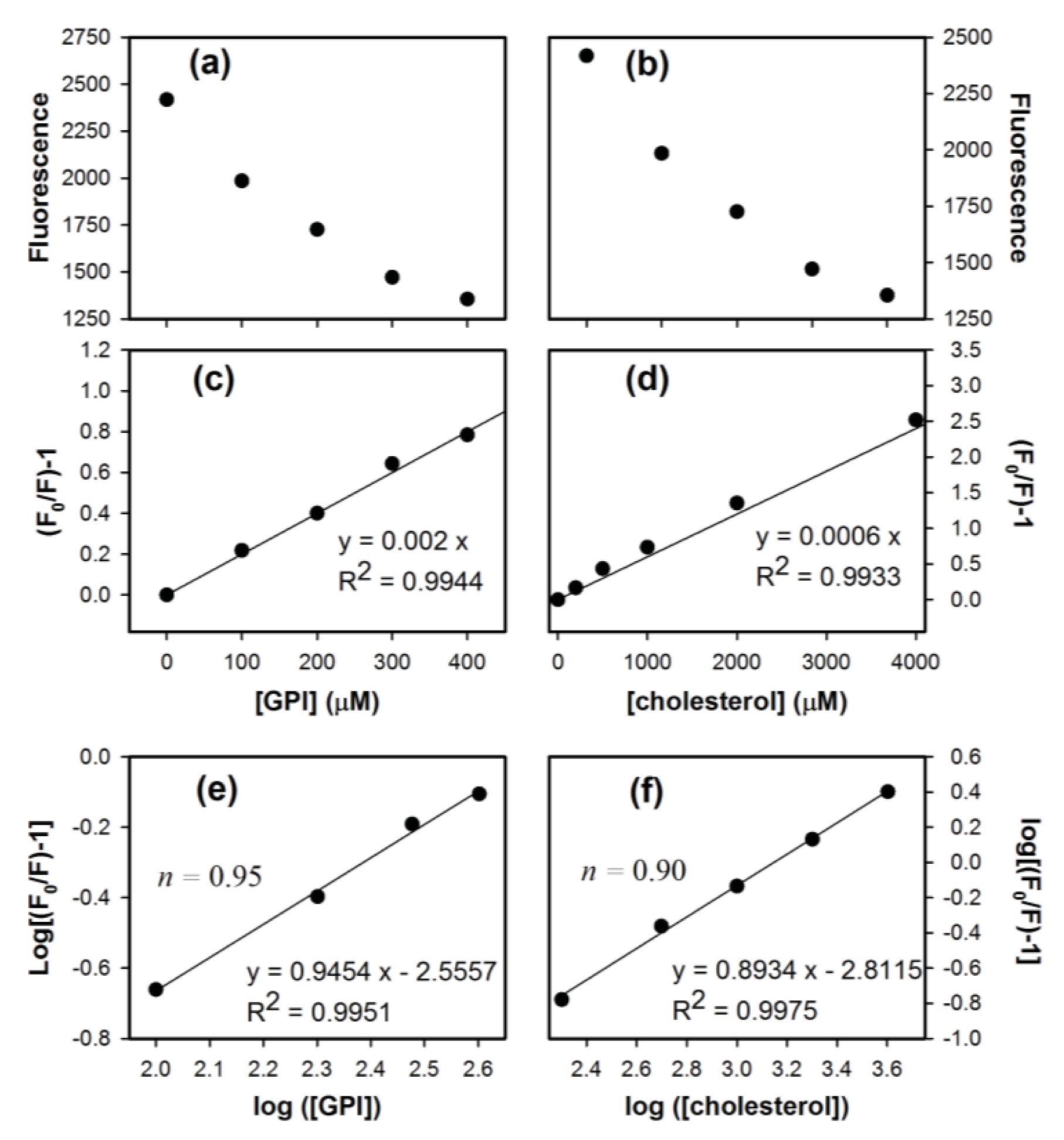
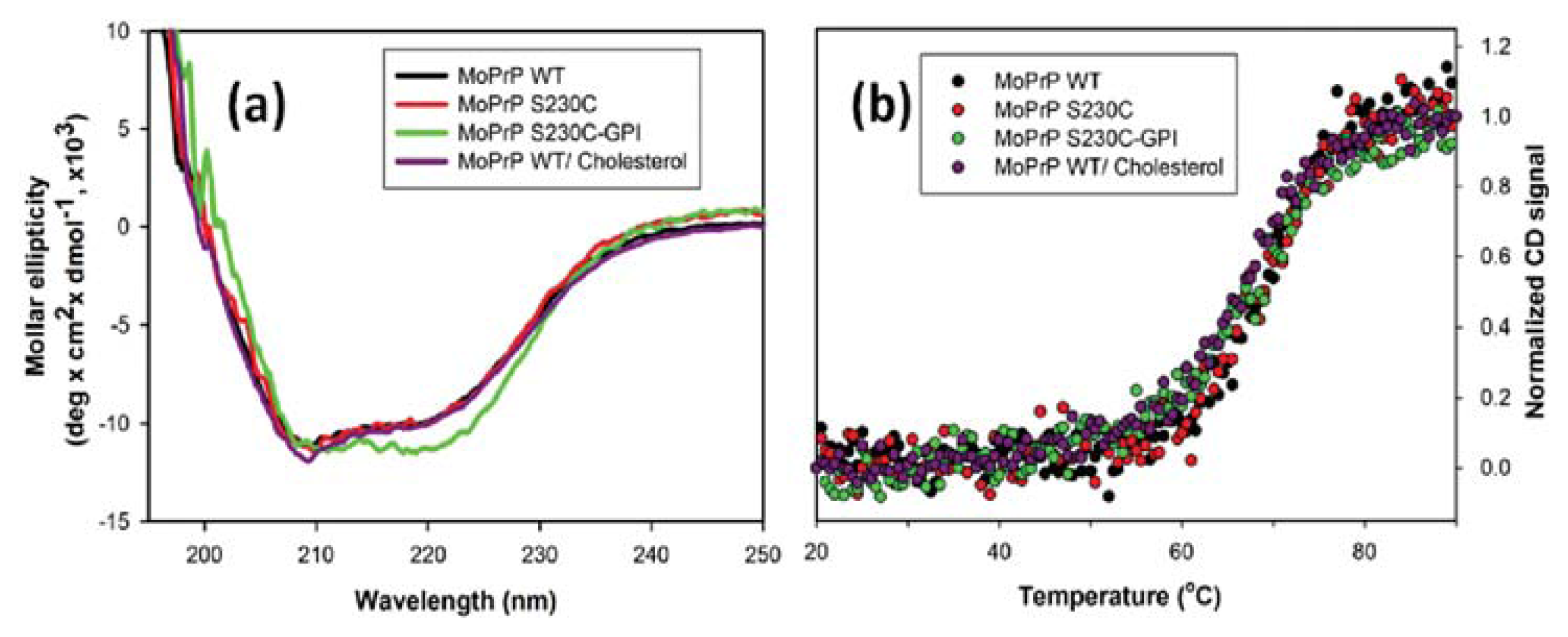
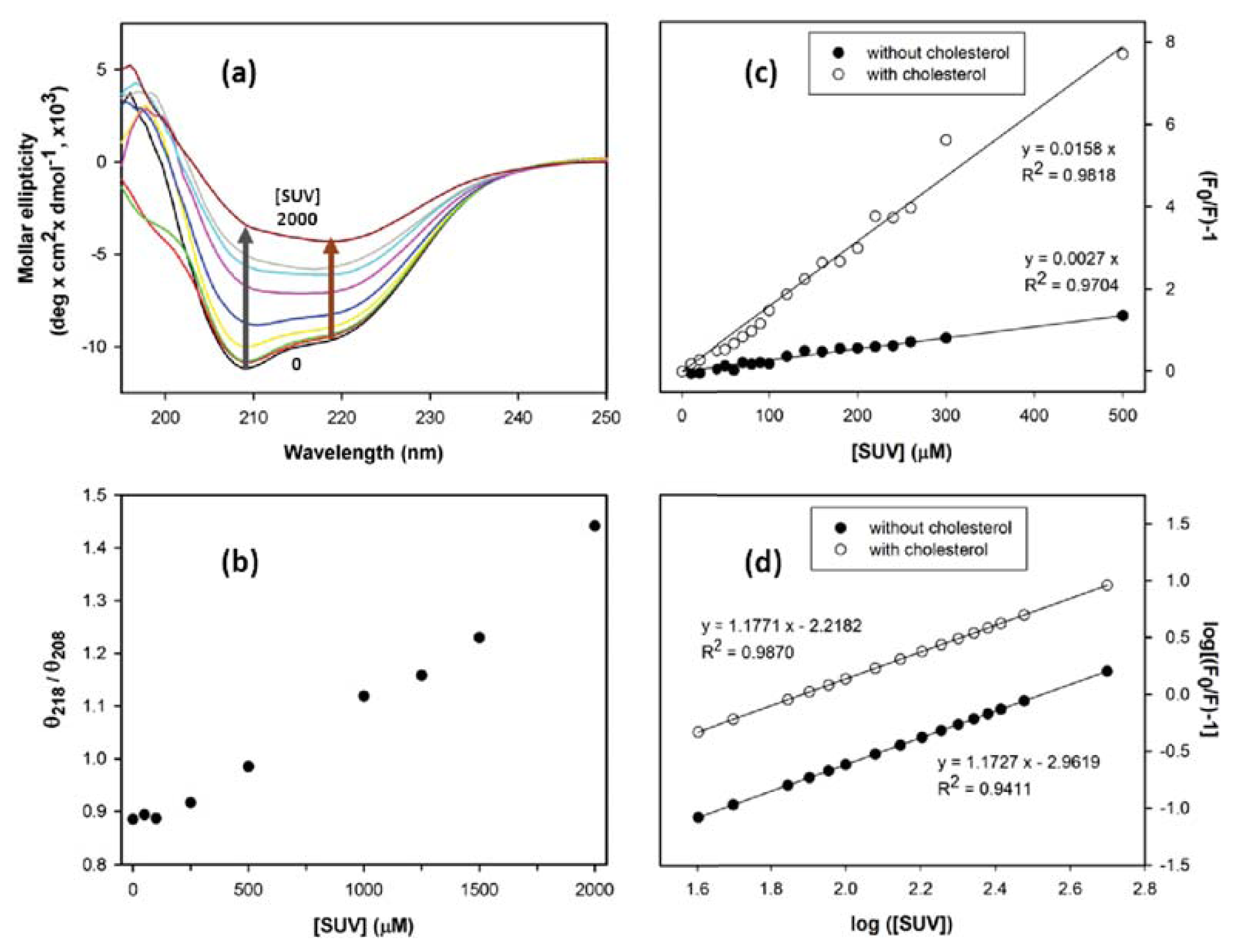
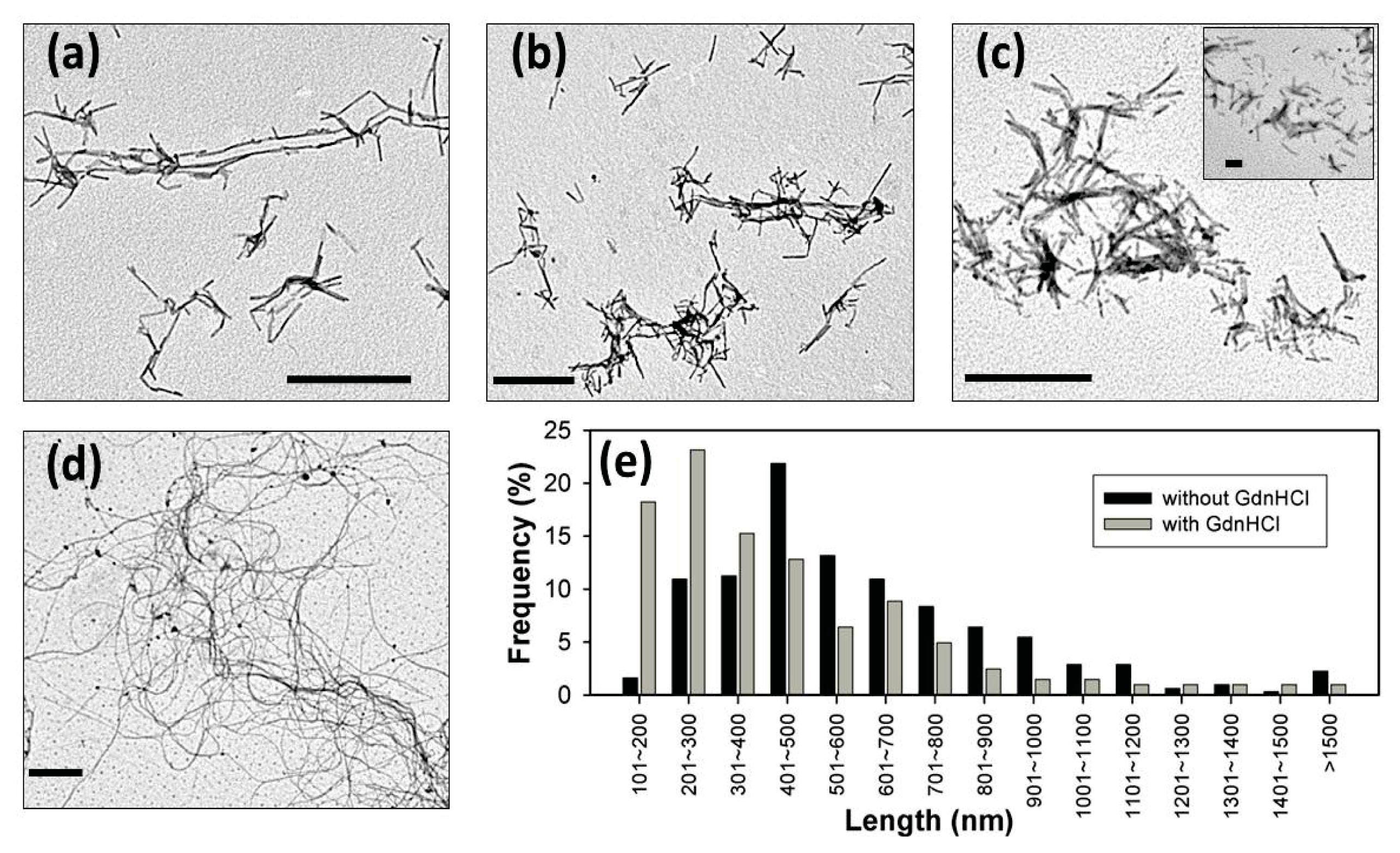
2. Results and Discussion
3. Experimental Section
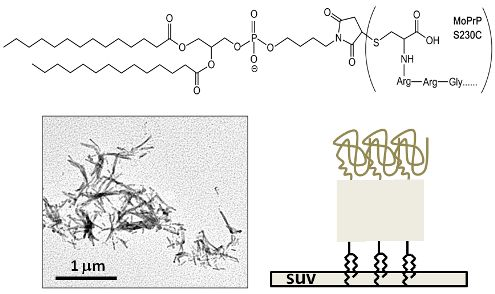
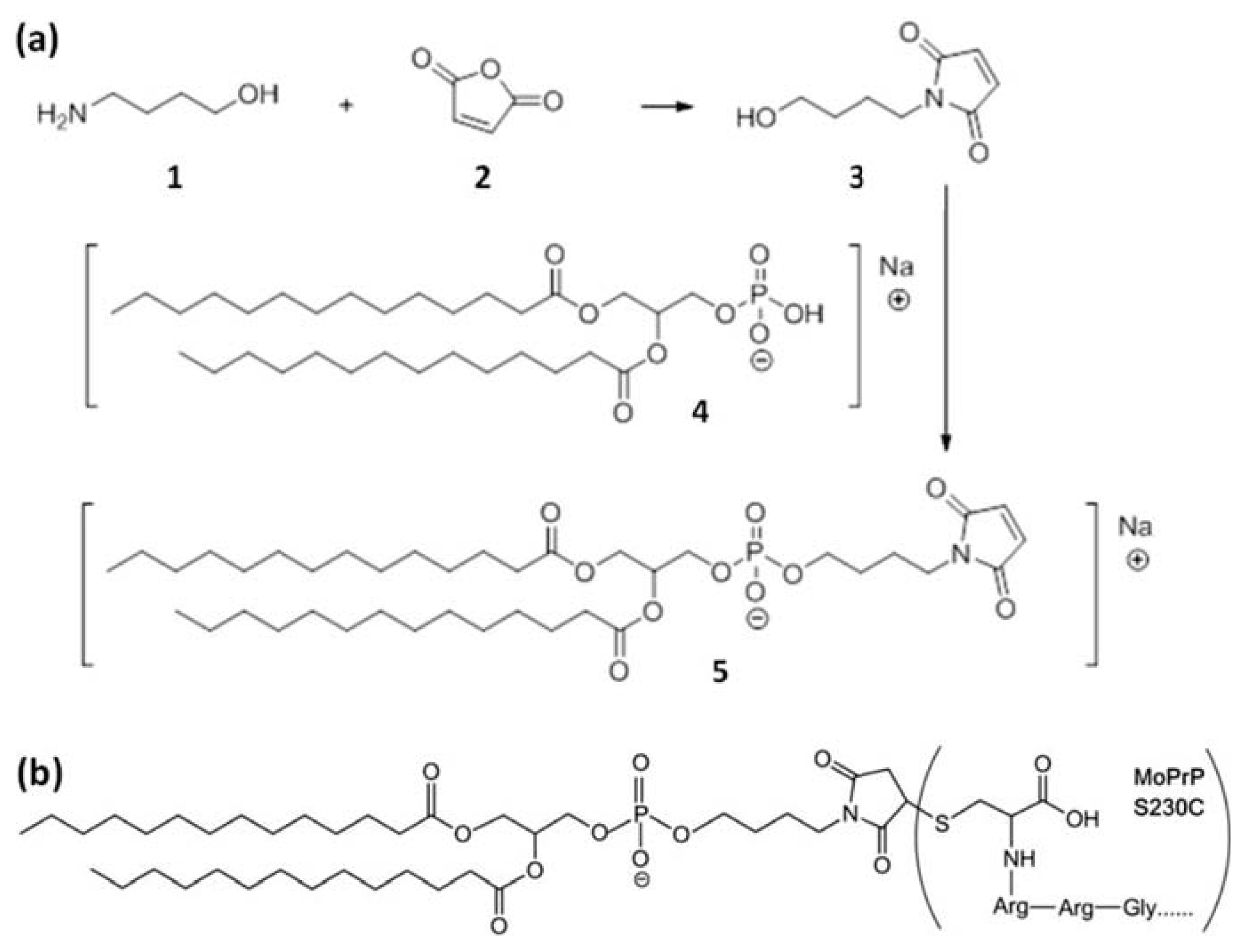
3.1. Synthesis of GPI-Analog
3.2. Covalent Linkage of MoPrP S230C and GPI Analog
3.3. Plasmid Construction
3.4. Protein Expression and Purification
3.5. Preparation of Small Unilamellar Lipid Vesicles (SUV)
3.6. Fluorescence Spectroscopy
3.7. Circular Dichroism Spectroscopy
3.8. Fibril Conversion
3.9. Transmission Electron Microscopy
3.10. Proteinase K Digestion
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Aguzzi, A.; Sigurdson, C.; Heikenwaelder, M. Molecular mechanisms of prion pathogenesis. Annu. Rev. Pathol 2008, 3, 11–40. [Google Scholar]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar]
- Vey, M.; Pilkuhn, S.; Wille, H.; Nixon, R.; DeArmond, S.J.; Smart, E.J.; Anderson, R.G.; Taraboulos, A.; Prusiner, S.B. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc. Natl. Acad. Sci. USA 1996, 93, 14945–14949. [Google Scholar]
- Hooper, N.M. Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae (review). Mol. Membr. Biol 1999, 16, 145–156. [Google Scholar]
- Taraboulos, A.; Scott, M.; Semenov, A.; Avrahami, D.; Laszlo, L.; Prusiner, S.B. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J. Cell Biol 1995, 129, 121–132. [Google Scholar]
- Kaneko, K.; Vey, M.; Scott, M.; Pilkuhn, S.; Cohen, F.E.; Prusiner, S.B. COOH-terminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform. Proc. Natl. Acad. Sci. USA 1997, 94, 2333–2338. [Google Scholar]
- Priola, S.A.; McNally, K.L. The role of the prion protein membrane anchor in prion infection. Prion 2009, 3, 134–138. [Google Scholar]
- Martin, S.; Parton, R.G. Caveolin, cholesterol, and lipid bodies. Semin. Cell Dev. Biol 2005, 16, 163–174. [Google Scholar]
- Miura, T.; Yoda, M.; Takaku, N.; Hirose, T.; Takeuchi, H. Clustered negative charges on the lipid membrane surface induce beta-sheet formation of prion protein fragment 106–126. Biochemistry 2007, 46, 11589–11597. [Google Scholar]
- Zhao, H.; Tuominen, E.K.; Kinnunen, P.K. Formation of amyloid fibers triggered by phosphatidylserine-containing membranes. Biochemistry 2004, 43, 10302–10307. [Google Scholar]
- Curtain, C.C.; Ali, F.E.; Smith, D.G.; Bush, A.I.; Masters, C.L.; Barnham, K.J. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-beta peptide with membrane lipid. J. Biol. Chem 2003, 278, 2977–2982. [Google Scholar]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005, 308, 1435–1439. [Google Scholar]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar]
- Swietnicki, W.; Morillas, M.; Chen, S.G.; Gambetti, P.; Surewicz, W.K. Aggregation and fibrillization of the recombinant human prion protein huPrP90–231. Biochemistry 2000, 39, 424–431. [Google Scholar]
- Bocharova, O.V.; Breydo, L.; Salnikov, V.V.; Gill, A.C.; Baskakov, I.V. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease. Protein Sci 2005, 14, 1222–1232. [Google Scholar]
- Zahn, R.; Liu, A.; Luhrs, T.; Riek, R.; von Schroetter, C.; Lopez Garcia, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wuthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar]
- Luo, J.C.; Wang, S.C.; Jian, W.B.; Chen, C.H.; Tang, J.L.; Lee, C.I. Formation of amyloid fibrils from beta-amylase. FEBS Lett 2012, 586, 680–685. [Google Scholar]
- Makarava, N.; Lee, C.I.; Ostapchenko, V.G.; Baskakov, I.V. Highly promiscuous nature of prion polymerization. J. Biol. Chem 2007, 282, 36704–36713. [Google Scholar]
- Breydo, L.; Sun, Y.; Makarava, N.; Lee, C.I.; Novitskaia, V.; Bocharova, O.; Kao, J.P.; Baskakov, I.V. Nonpolar substitution at the C-terminus of the prion protein, a mimic of the glycosylphosphatidylinositol anchor, partially impairs amyloid fibril formation. Biochemistry 2007, 46, 852–861. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed; Springer: New York, NY, USA, 2006. [Google Scholar]
- Elfrink, K.; Ollesch, J.; Stohr, J.; Willbold, D.; Riesner, D.; Gerwert, K. Structural changes of membrane-anchored native PrP(C). Proc. Natl. Acad. Sci. USA 2008, 105, 10815–10819. [Google Scholar]
- Morillas, M.; Swietnicki, W.; Gambetti, P.; Surewicz, W.K. Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem 1999, 274, 36859–36865. [Google Scholar]
- Tsiroulnikov, K.; Shchutskaya, Y.; Muronetz, V.; Chobert, J.M.; Haertle, T. Phospholipids influence the aggregation of recombinant ovine prions. From rapid extensive aggregation to amyloidogenic conversion. Biochim. Biophys. Acta 2009, 1794, 506–511. [Google Scholar]
- Wang, F.; Yang, F.; Hu, Y.; Wang, X.; Wang, X.; Jin, C.; Ma, J. Lipid interaction converts prion protein to a PrPSc-like proteinase K-resistant conformation under physiological conditions. Biochemistry 2007, 46, 7045–7053. [Google Scholar]
- Robinson, P.J.; Pinheiro, T.J. Phospholipid composition of membranes directs prions down alternative aggregation pathways. Biophys. J 2010, 98, 1520–1528. [Google Scholar]
- Baron, G.S.; Wehrly, K.; Dorward, D.W.; Chesebro, B.; Caughey, B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 2002, 21, 1031–1040. [Google Scholar]
- Mahal, S.P.; Jablonski, J.; Suponitsky-Kroyter, I.; Oelschlegel, A.M.; Herva, M.E.; Oldstone, M.; Weissmann, C. Propagation of RML prions in mice expressing PrP devoid of GPI anchor leads to formation of a novel, stable prion strain. PLoS Pathog 2012, 8, e1002746. [Google Scholar]
- Ikezawa, H. Glycosylphosphatidylinositol (GPI)-anchored proteins. Biol. Pharm. Bull 2002, 25, 409–417. [Google Scholar]
- Varma, R.; Mayor, S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature 1998, 394, 798–801. [Google Scholar]
- Friedrichson, T.; Kurzchalia, T.V. Microdomains of GPI-anchored proteins in living cells revealed by crosslinking. Nature 1998, 394, 802–805. [Google Scholar]
- Stulnig, T.M.; Berger, M.; Sigmund, T.; Stockinger, H.; Horejsi, V.; Waldhausl, W. Signal transduction via glycosyl phosphatidylinositol-anchored proteins in T cells is inhibited by lowering cellular cholesterol. J. Biol. Chem 1997, 272, 19242–19247. [Google Scholar]
- Simons, M.; Keller, P.; Dichgans, J.; Schulz, J.B. Cholesterol and Alzheimer’s disease: Is there a link? Neurology 2001, 57, 1089–1093. [Google Scholar]
- Matsuzaki, T.; Sasaki, K.; Hata, J.; Hirakawa, Y.; Fujimi, K.; Ninomiya, T.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; Iwaki, T. Association of Alzheimer disease pathology with abnormal lipid metabolism: The Hisayama study. Neurology 2011, 77, 1068–1075. [Google Scholar]
- Baskakov, I.V.; Legname, G.; Gryczynski, Z.; Prusiner, S.B. The peculiar nature of unfolding of the human prion protein. Protein Sci 2004, 13, 586–595. [Google Scholar]
- Lee, C.I.; Chang, N.Y. Characterizing the denatured state of human prion 121–230. Biophys. Chem 2010, 151, 86–90. [Google Scholar]
- Tang, J.L.; Wu, P.J.; Wang, S.C.; Lee, C.I. Insights into structural properties of denatured human prion 121–230 at melting temperature studied by replica exchange molecular dynamics. J. Phys. Chem. B 2012, 116, 3305–3312. [Google Scholar]
- Sunde, M.; Blake, C. The structure of amyloid fibrils by electron microscopy and X-ray diffraction. Adv. Protein Chem 1997, 50, 123–159. [Google Scholar]
- Coleman, L.; Bork, J.; Dunn, H. Reaction of primary aliphatic amines with maleic anhydride. J. Org. Chem 1959, 24, 135–136. [Google Scholar]
- Jiang, S.B.; Tala, S.R.; Lu, H.; Zou, P.; Avan, I.; Ibrahim, T.S.; Abo-Dya, N.E.; Abdelmajeid, A.; Debnath, A.K.; Katritzky, A.R. Design, synthesis, and biological activity of a novel series of 2,5-disubstituted furans/pyrroles as HIV-1 fusion inhibitors targeting gp41. Bioorg. Med. Chem. Lett 2011, 21, 6895–6898. [Google Scholar]
- Uragami, M.; Miyake, Y.; Tokutake, N.; Zhang, L.H.; Regen, S.L. Design and synthesis of phosphatidylcholine mimics and their mixing behavior with phosphatidylglycerol mimics in the fluid bilayer state. Langmuir 2000, 16, 8010–8015. [Google Scholar]
- Schagger, H. Tricine-SDS-PAGE. Nat. Protoc 2006, 1, 16–22. [Google Scholar]
- Bocharova, O.V.; Breydo, L.; Parfenov, A.S.; Salnikov, V.V.; Baskakov, I.V. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J. Mol. Biol 2005, 346, 645–659. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lin, S.-J.; Yu, K.-H.; Wu, J.-R.; Lee, C.-F.; Jheng, C.-P.; Chen, H.-R.; Lee, C.-I. Liberation of GPI-Anchored Prion from Phospholipids Accelerates Amyloidogenic Conversion. Int. J. Mol. Sci. 2013, 14, 17943-17957. https://doi.org/10.3390/ijms140917943
Lin S-J, Yu K-H, Wu J-R, Lee C-F, Jheng C-P, Chen H-R, Lee C-I. Liberation of GPI-Anchored Prion from Phospholipids Accelerates Amyloidogenic Conversion. International Journal of Molecular Sciences. 2013; 14(9):17943-17957. https://doi.org/10.3390/ijms140917943
Chicago/Turabian StyleLin, Shen-Jie, Kun-Hua Yu, Jhih-Ru Wu, Chin-Fa Lee, Cheng-Ping Jheng, Hau-Ren Chen, and Cheng-I Lee. 2013. "Liberation of GPI-Anchored Prion from Phospholipids Accelerates Amyloidogenic Conversion" International Journal of Molecular Sciences 14, no. 9: 17943-17957. https://doi.org/10.3390/ijms140917943