Mechanisms of Action of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Mesalazine in the Chemoprevention of Colorectal Cancer


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanisms of Action of NSAIDs in CRC Chemoprevention
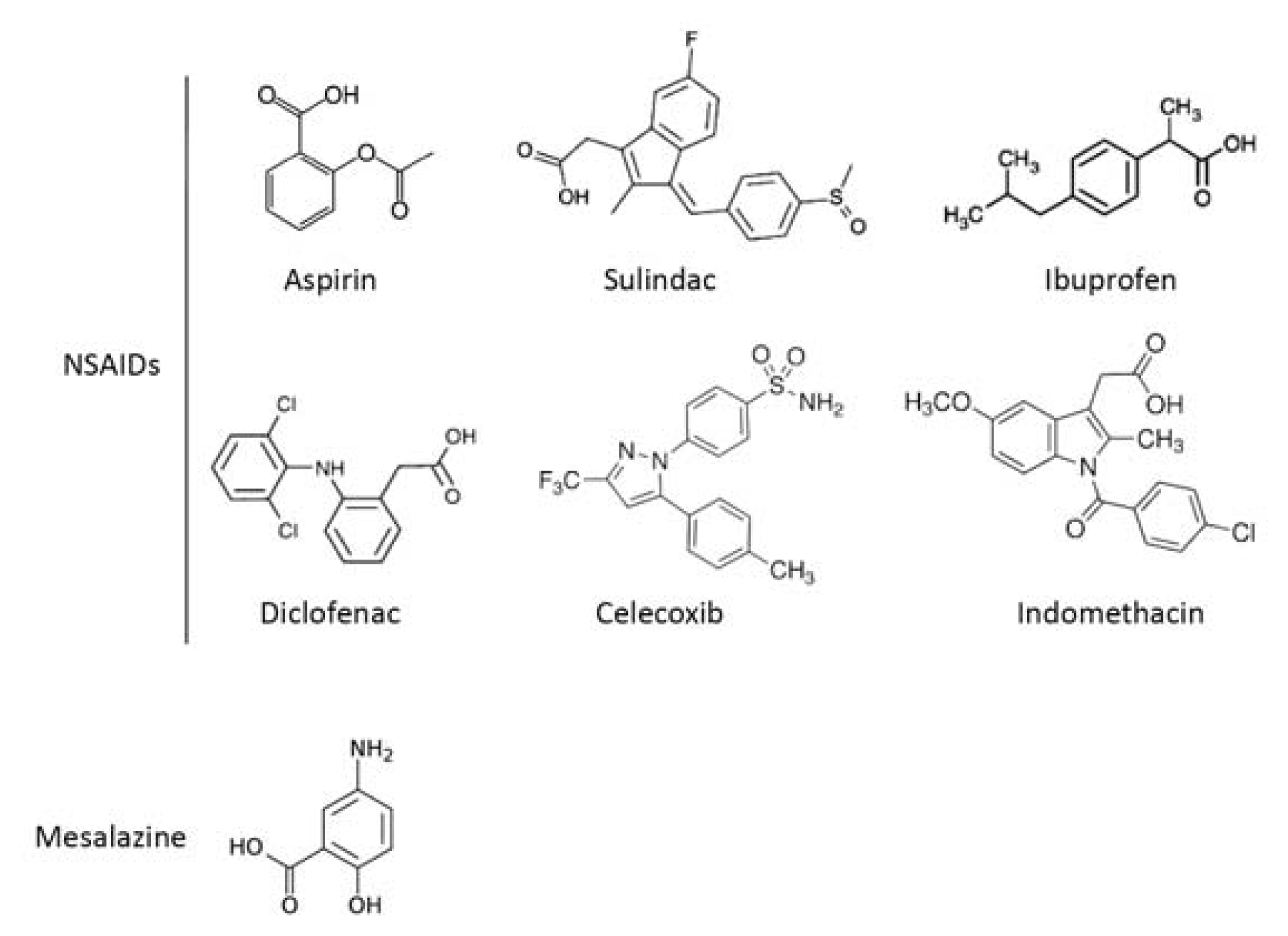
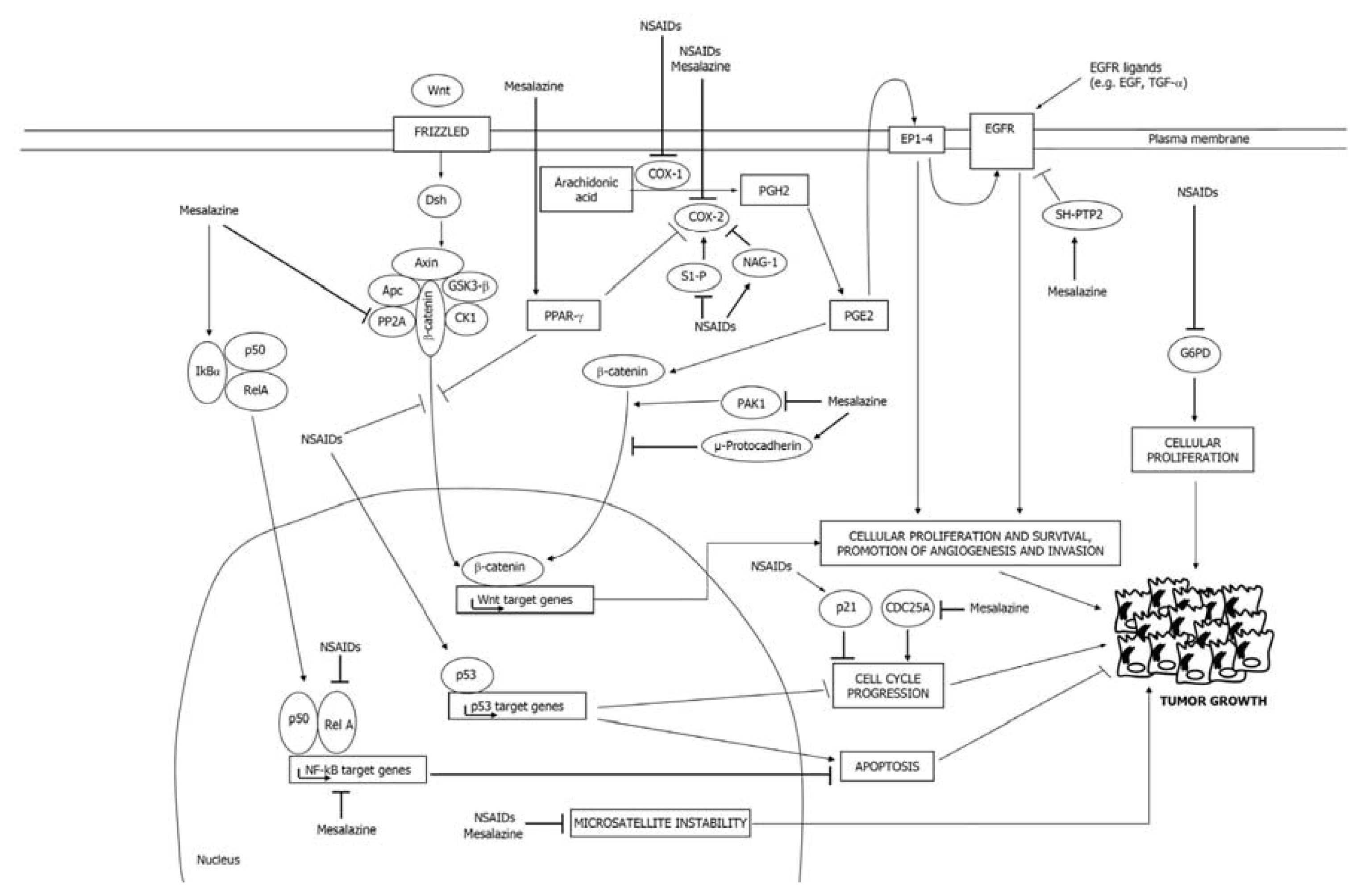
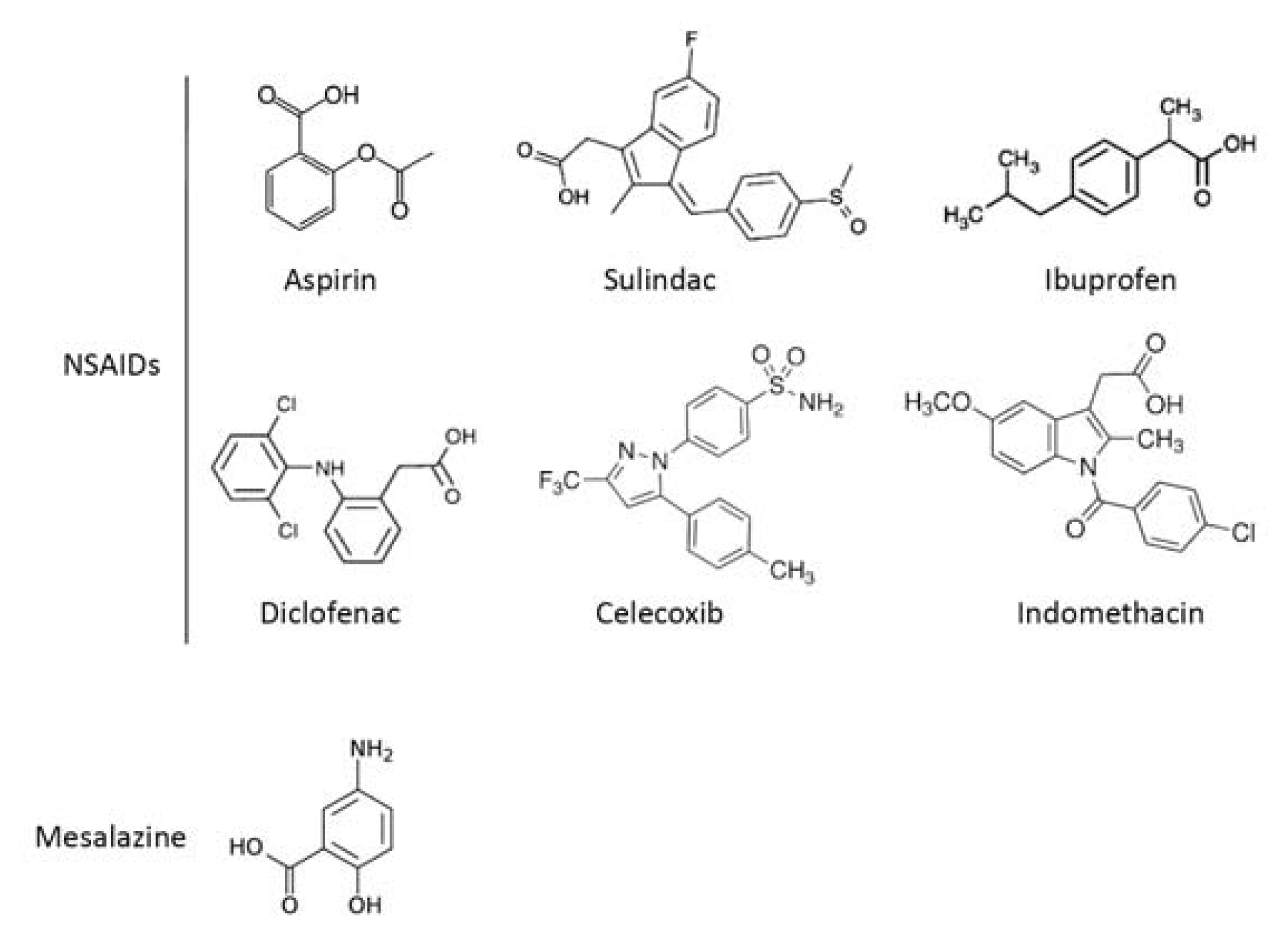
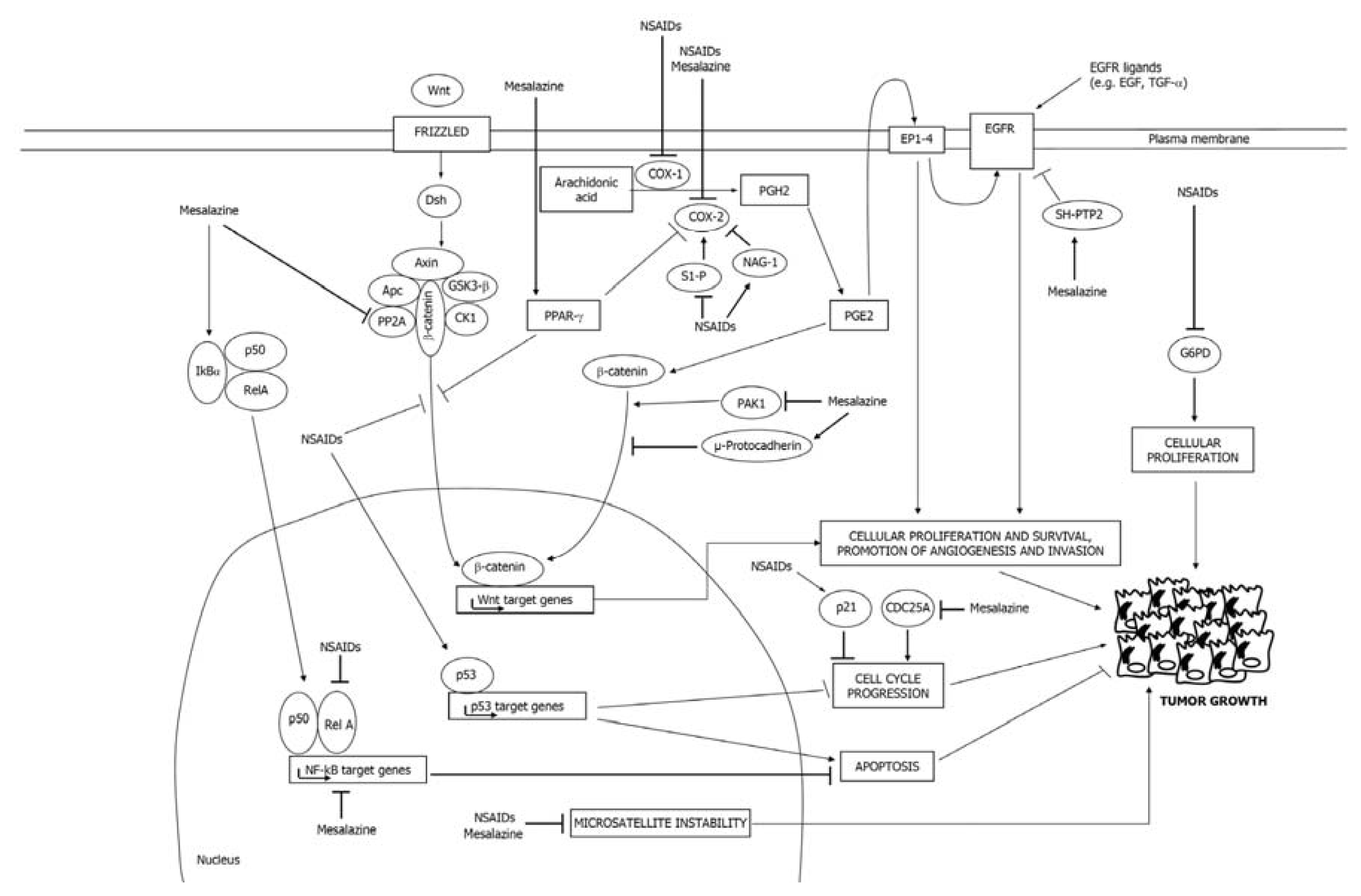
2.1. COX-Dependent Mechanisms
2.2. COX-Independent Mechanisms
3. Mechanisms of Action of Mesalazine in CRC Chemoprevention
3.1. Mesalazine Negatively Regulates the COX-2/PGE2 Axis
3.2. Mesalazine Inhibits EGFR, NF-κB and Wnt/β-Catenin Signaling
3.3. Mesalazine Activates PPAR-γ in CRC Cells
3.4. Mesalazine Modulates Cell Cycle-Related Proteins
3.5. Mesalazine Improves Replication Fidelity
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Center, M.M.; Jemal, A.; Smith, R.A.; Ward, E. Worldwide variations in colorectal cancer. CA Cancer J. Clin 2009, 59, 366–378. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar]
- Bertagnolli, M.M.; Eagle, C.J.; Zauber, A.G.; Redston, M.; Solomon, S.D.; Kim, K.; Tang, J.; Rosenstein, R.B.; Wittes, J.; Corle, D.; et al. Celecoxib for the prevention of sporadic colorectal adenomas. N. Engl. J. Med 2006, 355, 873–884. [Google Scholar]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar]
- Smalley, W.; Ray, W.A.; Daugherty, J.; Griffin, M.R. Use of nonsteroidal anti-inflammatory drugs and incidence of colorectal cancer: A population-based study. Arch. Intern. Med 1999, 159, 161–166. [Google Scholar]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar]
- Van Staa, T.P.; Card, T.; Logan, R.F.; Leufkens, H.G. 5-aminosalicylate use and colorectal cancer risk in inflammatory bowel disease: A large epidemiological study. Gut 2005, 54, 1573–1578. [Google Scholar]
- Rubin, D.T.; LoSavio, A.; Yadron, N.; Huo, D.; Hanauer, S.B. Aminosalicylate therapy in the prevention of dysplasia and colorectal cancer in ulcerative colitis. Clin. Gastroenterol. Hepatol 2006, 4, 1346–1350. [Google Scholar]
- Velayos, F.S.; Terdiman, J.P.; Walsh, J.M. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: A systematic review and metaanalysis of observational studies. Am. J. Gastroenterol 2005, 100, 1345–1353. [Google Scholar]
- Terdiman, J.P.; Steinbuch, M.; Blumentals, W.A.; Ullman, T.A.; Rubin, D.T. 5-aminosalicylic acid therapy and the risk of colorectal cancer among patients with inflammatory bowel disease. Inflamm. Bowel Dis 2007, 13, 367–371. [Google Scholar]
- Bernstein, C.N.; Nugent, Z.; Blanchard, J.F. 5-aminosalicylate is not chemoprophylactic for colorectal cancer in ibd: A population based study. Am. J. Gastroenterol 2011, 106, 731–736. [Google Scholar]
- Harris, R.E. Cyclooxygenase-2 (cox-2) and the inflammogenesis of cancer. Subcell. Biochem 2007, 42, 93–126. [Google Scholar]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenases 1 and 2. Annu. Rev. Pharmacol. Toxicol 1998, 38, 97–120. [Google Scholar]
- Chulada, P.C.; Thompson, M.B.; Mahler, J.F.; Doyle, C.M.; Gaul, B.W.; Lee, C.; Tiano, H.F.; Morham, S.G.; Smithies, O.; Langenbach, R. Genetic disruption of ptgs-1, as well as ptgs-2, reduces intestinal tumorigenesis in min mice. Cancer Res 2000, 60, 4705–4708. [Google Scholar]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; van de Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J 1998, 12, 1063–1073. [Google Scholar]
- Tsujii, M.; Kawano, S.; DuBois, R.N. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc. Natl. Acad. Sci. USA 1997, 94, 3336–3340. [Google Scholar]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar]
- Harris, R.E. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology 2009, 17, 55–67. [Google Scholar]
- Warner, T.D.; Giuliano, F.; Vojnovic, I.; Bukasa, A.; Mitchell, J.A.; Vane, J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: A full in vitro analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 7563–7568. [Google Scholar]
- Nash, G.F.; Turner, L.F.; Scully, M.F.; Kakkar, A.K. Platelets and cancer. Lancet Oncol 2002, 3, 425–430. [Google Scholar]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res 2012, 72, 726–735. [Google Scholar]
- Kawamori, T.; Osta, W.; Johnson, K.R.; Pettus, B.J.; Bielawski, J.; Tanaka, T.; Wargovich, M.J.; Reddy, B.S.; Hannun, Y.A.; Obeid, L.M.; et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J 2006, 20, 386–388. [Google Scholar]
- Ulrych, T.; Bohm, A.; Polzin, A.; Daum, G.; Nusing, R.M.; Geisslinger, G.; Hohlfeld, T.; Schror, K.; Rauch, B.H. Release of sphingosine-1-phosphate from human platelets is dependent on thromboxane formation. J. Thromb. Haemost 2011, 9, 790–798. [Google Scholar]
- Eling, T.E.; Baek, S.J.; Shim, M.; Lee, C.H. Nsaid activated gene (nag-1), a modulator of tumorigenesis. J. Biochem. Mol. Biol 2006, 39, 649–655. [Google Scholar]
- Kim, K.S.; Baek, S.J.; Flake, G.P.; Loftin, C.D.; Calvo, B.F.; Eling, T.E. Expression and regulation of nonsteroidal anti-inflammatory drug-activated gene (nag-1) in human and mouse tissue. Gastroenterology 2002, 122, 1388–1398. [Google Scholar]
- Baek, S.J.; Kim, K.S.; Nixon, J.B.; Wilson, L.C.; Eling, T.E. Cyclooxygenase inhibitors regulate the expression of a tgf-beta superfamily member that has proapoptotic and antitumorigenic activities. Mol. Pharmacol 2001, 59, 901–908. [Google Scholar]
- Iguchi, G.; Chrysovergis, K.; Lee, S.H.; Baek, S.J.; Langenbach, R.; Eling, T.E. A reciprocal relationship exists between non-steroidal anti-inflammatory drug-activated gene-1 (nag-1) and cyclooxygenase-2. Cancer Lett 2009, 282, 152–158. [Google Scholar]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin-independent pathway. Biochem. Pharmacol 1996, 52, 237–245. [Google Scholar]
- Smith, M.L.; Hawcroft, G.; Hull, M.A. The effect of non-steroidal anti-inflammatory drugs on human colorectal cancer cells: Evidence of different mechanisms of action. Eur. J. Cancer 2000, 36, 664–674. [Google Scholar]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar]
- Din, F.V.; Stark, L.A.; Dunlop, M.G. Aspirin-induced nuclear translocation of nfkappab and apoptosis in colorectal cancer is independent of p53 status and DNA mismatch repair proficiency. Br. J. Cancer 2005, 92, 1137–1143. [Google Scholar]
- Stark, L.A.; Reid, K.; Sansom, O.J.; Din, F.V.; Guichard, S.; Mayer, I.; Jodrell, D.I.; Clarke, A.R.; Dunlop, M.G. Aspirin activates the NF-kappaB signalling pathway and induces apoptosis in intestinal neoplasia in two in vivo models of human colorectal cancer. Carcinogenesis 2007, 28, 968–976. [Google Scholar]
- Li, X.; Gao, L.; Cui, Q.; Gary, B.D.; Dyess, D.L.; Taylor, W.; Shevde, L.A.; Samant, R.S.; Dean-Colomb, W.; Piazza, G.A.; et al. Sulindac inhibits tumor cell invasion by suppressing NF-kappaB-mediated transcription of micrornas. Oncogene 2012, 31, 4979–4986. [Google Scholar]
- Loveridge, C.J.; MacDonald, A.D.; Thoms, H.C.; Dunlop, M.G.; Stark, L.A. The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the rela(p65) subunit of NF-kappaB. Oncogene 2008, 27, 2648–2655. [Google Scholar]
- Bienz, M.; Clevers, H. Linking colorectal cancer to wnt signaling. Cell 2000, 103, 311–320. [Google Scholar]
- Bos, C.L.; Kodach, L.L.; van den Brink, G.R.; Diks, S.H.; van Santen, M.M.; Richel, D.J.; Peppelenbosch, M.P.; Hardwick, J.C. Effect of aspirin on the wnt/beta-catenin pathway is mediated via protein phosphatase 2a. Oncogene 2006, 25, 6447–6456. [Google Scholar]
- Maier, T.J.; Janssen, A.; Schmidt, R.; Geisslinger, G.; Grosch, S. Targeting the beta-catenin/apc pathway: A novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J 2005, 19, 1353–1355. [Google Scholar]
- Gardner, S.H.; Hawcroft, G.; Hull, M.A. Effect of nonsteroidal anti-inflammatory drugs on beta-catenin protein levels and catenin-related transcription in human colorectal cancer cells. Br. J. Cancer 2004, 91, 153–163. [Google Scholar]
- Rice, P.L.; Kelloff, J.; Sullivan, H.; Driggers, L.J.; Beard, K.S.; Kuwada, S.; Piazza, G.; Ahnen, D.J. Sulindac metabolites induce caspase- and proteasome-dependent degradation of beta-catenin protein in human colon cancer cells. Mol. Cancer Ther 2003, 2, 885–892. [Google Scholar]
- Li, N.; Xi, Y.; Tinsley, H.N.; Gurpinar, E.; Gary, B.D.; Zhu, B.; Li, Y.; Chen, X.; Keeton, A.B.; Abadi, A.H.; et al. Sulindac selectively inhibits colon tumor cell growth by activating the cgmp/pkg pathway to suppress wnt/beta-catenin signaling. Mol. Cancer Ther. 2013, in press. [Google Scholar]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Alfonso, L.F.; Srivenugopal, K.S.; Arumugam, T.V.; Abbruscato, T.J.; Weidanz, J.A.; Bhat, G.J. Aspirin inhibits camptothecin-induced p21cip1 levels and potentiates apoptosis in human breast cancer cells. Int. J. Oncol 2009, 34, 597–608. [Google Scholar]
- Marimuthu, S.; Chivukula, R.S.; Alfonso, L.F.; Moridani, M.; Hagen, F.K.; Bhat, G.J. Aspirin acetylates multiple cellular proteins in hct-116 colon cancer cells: Identification of novel targets. Int. J. Oncol 2011, 39, 1273–1283. [Google Scholar]
- Castells, A.; Paya, A.; Alenda, C.; Rodriguez-Moranta, F.; Agrelo, R.; Andreu, M.; Pinol, V.; Castellvi-Bel, S.; Jover, R.; Llor, X.; et al. Cyclooxygenase 2 expression in colorectal cancer with DNA mismatch repair deficiency. Clin. Cancer Res 2006, 12, 1686–1692. [Google Scholar]
- Ruschoff, J.; Wallinger, S.; Dietmaier, W.; Bocker, T.; Brockhoff, G.; Hofstadter, F.; Fishel, R. Aspirin suppresses the mutator phenotype associated with hereditary nonpolyposis colorectal cancer by genetic selection. Proc. Natl. Acad. Sci. USA 1998, 95, 11301–11306. [Google Scholar]
- Hsu, C.S.; Li, Y. Aspirin potently inhibits oxidative DNA strand breaks: Implications for cancer chemoprevention. Biochem. Biophys. Res. Commun 2002, 293, 705–709. [Google Scholar]
- Goel, A.; Chang, D.K.; Ricciardiello, L.; Gasche, C.; Boland, C.R. A novel mechanism for aspirin-mediated growth inhibition of human colon cancer cells. Clin. Cancer Res 2003, 9, 383–390. [Google Scholar]
- McIlhatton, M.A.; Tyler, J.; Kerepesi, L.A.; Bocker-Edmonston, T.; Kucherlapati, M.H.; Edelmann, W.; Kucherlapati, R.; Kopelovich, L.; Fishel, R. Aspirin and low-dose nitric oxide-donating aspirin increase life span in a lynch syndrome mouse model. Cancer Prev. Res 2011, 4, 684–693. [Google Scholar]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar]
- Luciani, M.G.; Campregher, C.; Gasche, C. Aspirin blocks proliferation in colon cells by inducing a g1 arrest and apoptosis through activation of the checkpoint kinase atm. Carcinogenesis 2007, 28, 2207–2217. [Google Scholar]
- Collier, H.O.; Francis, A.A.; McDonald-Gibson, W.J.; Saeed, S.A. Inhibition of prostaglandin biosynthesis by sulphasalazine and its metabolites. Prostaglandins 1976, 11, 219–225. [Google Scholar]
- Sharon, P.; Ligumsky, M.; Rachmilewitz, D.; Zor, U. Role of prostaglandins in ulcerative colitis. Enhanced production during active disease and inhibition by sulfasalazine. Gastroenterology 1978, 75, 638–640. [Google Scholar]
- Stolfi, C.; Fina, D.; Caruso, R.; Caprioli, F.; Sarra, M.; Fantini, M.C.; Rizzo, A.; Pallone, F.; Monteleone, G. Cyclooxygenase-2-dependent and -independent inhibition of proliferation of colon cancer cells by 5-aminosalicylic acid. Biochem. Pharmacol 2008, 75, 668–676. [Google Scholar]
- Clapper, M.L.; Gary, M.A.; Coudry, R.A.; Litwin, S.; Chang, W.C.; Devarajan, K.; Lubet, R.A.; Cooper, H.S. 5-aminosalicylic acid inhibits colitis-associated colorectal dysplasias in the mouse model of azoxymethane/dextran sulfate sodium-induced colitis. Inflamm. Bowel Dis 2008, 14, 1341–1347. [Google Scholar]
- Bradley, S.J.; Garfinkle, G.; Walker, E.; Salem, R.; Chen, L.B.; Steele, G., Jr. Increased expression of the epidermal growth factor receptor on human colon carcinoma cells. Arch. Surg. 1986, 121, 1242–1247. [Google Scholar]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell. Res 2003, 284, 31–53. [Google Scholar]
- Monteleone, G.; Franchi, L.; Fina, D.; Caruso, R.; Vavassori, P.; Monteleone, I.; Calabrese, E.; Naccari, G.C.; Bellinvia, S.; Testi, R.; et al. Silencing of sh-ptp2 defines a crucial role in the inactivation of epidermal growth factor receptor by 5-aminosalicylic acid in colon cancer cells. Cell Death Differ 2006, 13, 202–211. [Google Scholar]
- Kaiser, G.C.; Yan, F.; Polk, D.B. Mesalamine blocks tumor necrosis factor growth inhibition and nuclear factor kappaB activation in mouse colonocytes. Gastroenterology 1999, 116, 602–609. [Google Scholar]
- Egan, L.J.; Mays, D.C.; Huntoon, C.J.; Bell, M.P.; Pike, M.G.; Sandborn, W.J.; Lipsky, J.J.; McKean, D.J. Inhibition of interleukin-1-stimulated NF-kappaB rela/p65 phosphorylation by mesalamine is accompanied by decreased transcriptional activity. J. Biol. Chem 1999, 274, 26448–26453. [Google Scholar]
- Bos, C.L.; Diks, S.H.; Hardwick, J.C.; Walburg, K.V.; Peppelenbosch, M.P.; Richel, D.J. Protein phosphatase 2a is required for mesalazine-dependent inhibition of wnt/beta-catenin pathway activity. Carcinogenesis 2006, 27, 2371–2382. [Google Scholar]
- Parenti, S.; Ferrarini, F.; Zini, R.; Montanari, M.; Losi, L.; Canovi, B.; Ferrari, S.; Grande, A. Mesalazine inhibits the beta-catenin signalling pathway acting through the upregulation of mu-protocadherin gene in colo-rectal cancer cells. Aliment. Pharmacol. Ther 2010, 31, 108–119. [Google Scholar]
- Munding, J.; Ziebarth, W.; Pox, C.P.; Ladigan, S.; Reiser, M.; Huppe, D.; Brand, L.; Schmiegel, W.; Tannapfel, A.; Reinacher-Schick, A.C. The influence of 5-aminosalicylic acid on the progression of colorectal adenomas via the beta-catenin signaling pathway. Carcinogenesis 2012, 33, 637–643. [Google Scholar]
- Khare, V.; Lyakhovich, A.; Dammann, K.; Lang, M.; Borgmann, M.; Tichy, B.; Pospisilova, S.; Luciani, G.; Campregher, C.; Evstatiev, R.; et al. Mesalamine modulates intercellular adhesion through inhibition of p-21 activated kinase-1. Biochem. Pharmacol 2013, 85, 234–244. [Google Scholar]
- Matthiessen, M.W.; Pedersen, G.; Albrektsen, T.; Adamsen, S.; Fleckner, J.; Brynskov, J. Peroxisome proliferator-activated receptor expression and activation in normal human colonic epithelial cells and tubular adenomas. Scand. J. Gastroenterol 2005, 40, 198–205. [Google Scholar]
- Shimada, T.; Kojima, K.; Yoshiura, K.; Hiraishi, H.; Terano, A. Characteristics of the peroxisome proliferator activated receptor gamma (ppargamma) ligand induced apoptosis in colon cancer cells. Gut 2002, 50, 658–664. [Google Scholar]
- Osawa, E.; Nakajima, A.; Wada, K.; Ishimine, S.; Fujisawa, N.; Kawamori, T.; Matsuhashi, N.; Kadowaki, T.; Ochiai, M.; Sekihara, H.; et al. Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology 2003, 124, 361–367. [Google Scholar]
- Tanaka, T.; Kohno, H.; Yoshitani, S.; Takashima, S.; Okumura, A.; Murakami, A.; Hosokawa, M. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res 2001, 61, 2424–2428. [Google Scholar]
- Rousseaux, C.; Lefebvre, B.; Dubuquoy, L.; Lefebvre, P.; Romano, O.; Auwerx, J.; Metzger, D.; Wahli, W.; Desvergne, B.; Naccari, G.C.; et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J. Exp. Med 2005, 201, 1205–1215. [Google Scholar]
- Desreumaux, P.; Ghosh, S. Review article: Mode of action and delivery of 5-aminosalicylic acid-new evidence. Aliment. Pharmacol. Ther 2006, 24, S2–S9. [Google Scholar]
- Schwab, M.; Reynders, V.; Loitsch, S.; Shastri, Y.M.; Steinhilber, D.; Schroder, O.; Stein, J. PPARgamma is involved in mesalazine-mediated induction of apoptosis and inhibition of cell growth in colon cancer cells. Carcinogenesis 2008, 29, 1407–1414. [Google Scholar]
- Stolfi, C.; Fina, D.; Caruso, R.; Caprioli, F.; Fantini, M.C.; Rizzo, A.; Sarra, M.; Pallone, F.; Monteleone, G. Mesalazine negatively regulates cdc25a protein expression and promotes accumulation of colon cancer cells in s phase. Carcinogenesis 2008, 29, 1258–1266. [Google Scholar]
- Luciani, M.G.; Campregher, C.; Fortune, J.M.; Kunkel, T.A.; Gasche, C. 5-asa affects cell cycle progression in colorectal cells by reversibly activating a replication checkpoint. Gastroenterology 2007, 132, 221–235. [Google Scholar]
- Baan, B.; Dihal, A.A.; Hoff, E.; Bos, C.L.; Voorneveld, P.W.; Koelink, P.J.; Wildenberg, M.E.; Muncan, V.; Heijmans, J.; Verspaget, H.W.; et al. 5-aminosalicylic acid inhibits cell cycle progression in a phospholipase D dependent manner in colorectal cancer. Gut 2011, 61, 1708–1715. [Google Scholar]
- Koelink, P.J.; Mieremet-Ooms, M.A.; Corver, W.E.; Wolanin, K.; Hommes, D.W.; Lamers, C.B.; Verspaget, H.W. 5-aminosalicylic acid interferes in the cell cycle of colorectal cancer cells and induces cell death modes. Inflamm. Bowel Dis 2010, 16, 379–389. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar]
- Gasche, C.; Goel, A.; Natarajan, L.; Boland, C.R. Mesalazine improves replication fidelity in cultured colorectal cells. Cancer Res 2005, 65, 3993–3997. [Google Scholar]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar]
- Campregher, C.; Honeder, C.; Chung, H.; Carethers, J.M.; Gasche, C. Mesalazine reduces mutations in transforming growth factor beta receptor ii and activin type ii receptor by improvement of replication fidelity in mononucleotide repeats. Clin. Cancer Res 2010, 16, 1950–1956. [Google Scholar]
- Tinsley, H.N.; Piazza, G.A. Novel therapeutics: Nsaids, derivatives, and phosphodiesterases. Curr. Colorectal Cancer Rep 2012, 8, 325–330. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stolfi, C.; De Simone, V.; Pallone, F.; Monteleone, G. Mechanisms of Action of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Mesalazine in the Chemoprevention of Colorectal Cancer. Int. J. Mol. Sci. 2013, 14, 17972-17985. https://doi.org/10.3390/ijms140917972
Stolfi C, De Simone V, Pallone F, Monteleone G. Mechanisms of Action of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Mesalazine in the Chemoprevention of Colorectal Cancer. International Journal of Molecular Sciences. 2013; 14(9):17972-17985. https://doi.org/10.3390/ijms140917972
Chicago/Turabian StyleStolfi, Carmine, Veronica De Simone, Francesco Pallone, and Giovanni Monteleone. 2013. "Mechanisms of Action of Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Mesalazine in the Chemoprevention of Colorectal Cancer" International Journal of Molecular Sciences 14, no. 9: 17972-17985. https://doi.org/10.3390/ijms140917972