2.1. Relation between Crystalline Packing of the Monomer and Self-Organization of the Polymer in Bulk
A study of poly[oxy(2,4,4,6,8,8-hexaorganocyclotetrasiloxane-2,6-diyl)]s with different substituents showed that the formation of the mesomorphic state with the 2D type of packing and the existence of the region of this state depended on the intermolecular interactions of the side groups [
4].
Table 1 lists the phase transition temperatures of the CL polymers with the structural Type
I bearing different alkyl substituents. The narrowest existence region of the mesomorphic state was found for the mainly syndiotactic polymer with methyl substituents (syndiotactic
I-
Me). An atactic Polymer
I-
Me does not form the mesophase. In contrast to the atactic Polymer
I-
Me, atactic poly[oxy(2,4,4,6,6,8,10,10,12,12-decamethylcyclohexasiloxane-2,8-diyl)] (
II-
Me) forms the mesomorphic phase in a wide temperature interval; the introduction of phenyl substituents leads to an increase in the isotropization temperature
Ti [
13]. It should be noted that the tacticity of the polymer chain in the CL POS with the structural Type
II has little effect on the appearance of the mesophase with the 1D packing type [
2,
4]. Yet another property of CL POS with the structural Type
II is noteworthy: Namely, this is the effect of the molecular weight on the change in
Ti [
4,
5]. Unlike linear polydiorganosiloxanes, the mesomorphic state in CL POS
II appears at much lower degrees of polymerization (
n = 3–5) [
14–
16].
Table 2 presents the phase transition temperatures of the CL POS with the structural Type
II. Thus, CL POS with symmetric chain units of the structural Types
I and
II form the mesomorphic state. Comparing their ability to form the mesophase with that of linear polydiethylsiloxane, one can state that the major contribution to self-organization of the macromolecules of CL polymers comes from the cyclic fragments.
To assess the influence of oxygen atoms in the unit of CL PMS on the ability to self-organize, the bridging oxygen atoms between the cyclosiloxane moieties in I-Me, I-Et, and II-Me were replaced by some other species.
Cyclolinear polyorganocarbosiloxanes with ethylene groups instead of oxygen atoms >SiMeOMeSi< as bridges between rings were synthesized by the polyaddition reaction of dihydridocycloorganotetra(hexa,octa)siloxanes with divinylcycloorganotetra(hexa)siloxanes bearing methyl and ethyl substituents in the presence of platinum complex catalysts (the Karstedt and Speier catalysts, dicyclopentadienylplatinum dichloride) and their reduced forms following
Scheme 1 [
17].
1H and
13C nuclear magnetic resonance (NMR) studies revealed that the polyaddition reaction obeys Farmer’s rule and that the degree of formation of CH
2CH
2 bridges between the rings is higher than 95% [
17]. Using differential scanning calorimetry (DSC) and X-ray analysis, it was found that the CL POCS with the hexaorganocyclotetrasiloxane moiety in the chain unit bearing methyl (
III-
Me) and ethyl (
III-
Et) substituents were crystalline and amorphous, respectively. Compound
IV-
Me,
i.e., the CL POCS with the decamethylcyclohexasiloxane fragment of the chain unit was also amorphous in the temperature interval studied.
Tables 1 and
2 list the transition temperatures of the CL POCS with ethylene bridges between the cyclotetra- (
III-
Me and
III-
Et) and cyclohexasiloxane moieties (
IV-
Me and
IV-
Et), as well as that of the copolymer with alternating cyclohexa- and cyclooctasiloxane moieties (
V-
Me). All samples except
IV-
Et exhibit no mesomorphic properties.
Thus, the ability of CL POCS with methyl substituents to self-organize is suppressed upon replacement of the oxygen bridge by the CH
2CH
2 unit. Attempts to change the ratio of the number of SiO groups to the number of SiCH
2 groups (hereafter, the SiO/SiCH
2 ratio) in the main chain by varying the ring size from cyclotetrasiloxane to cyclooctasiloxane did not lead to recovery of the ability to self-organize with the formation of a mesomorphic state. The ability to self-organize in a wide temperature range appears only upon introducing ethyl substituents into the CL POCS with the Structure
IV [
17], similarly to the case for the CL POS
I-
Et (
Table 1). The close values of
Tg,
Ti, and the existence regions of the mesomorphic state of the CL POS
I-
Et and CL POCS
IV-
Et are noteworthy. This means that the contribution of structural changes in the main chain becomes less significant owing to strengthening of intermolecular interactions involving organic substituents. The X-ray diffraction pattern of Polymer
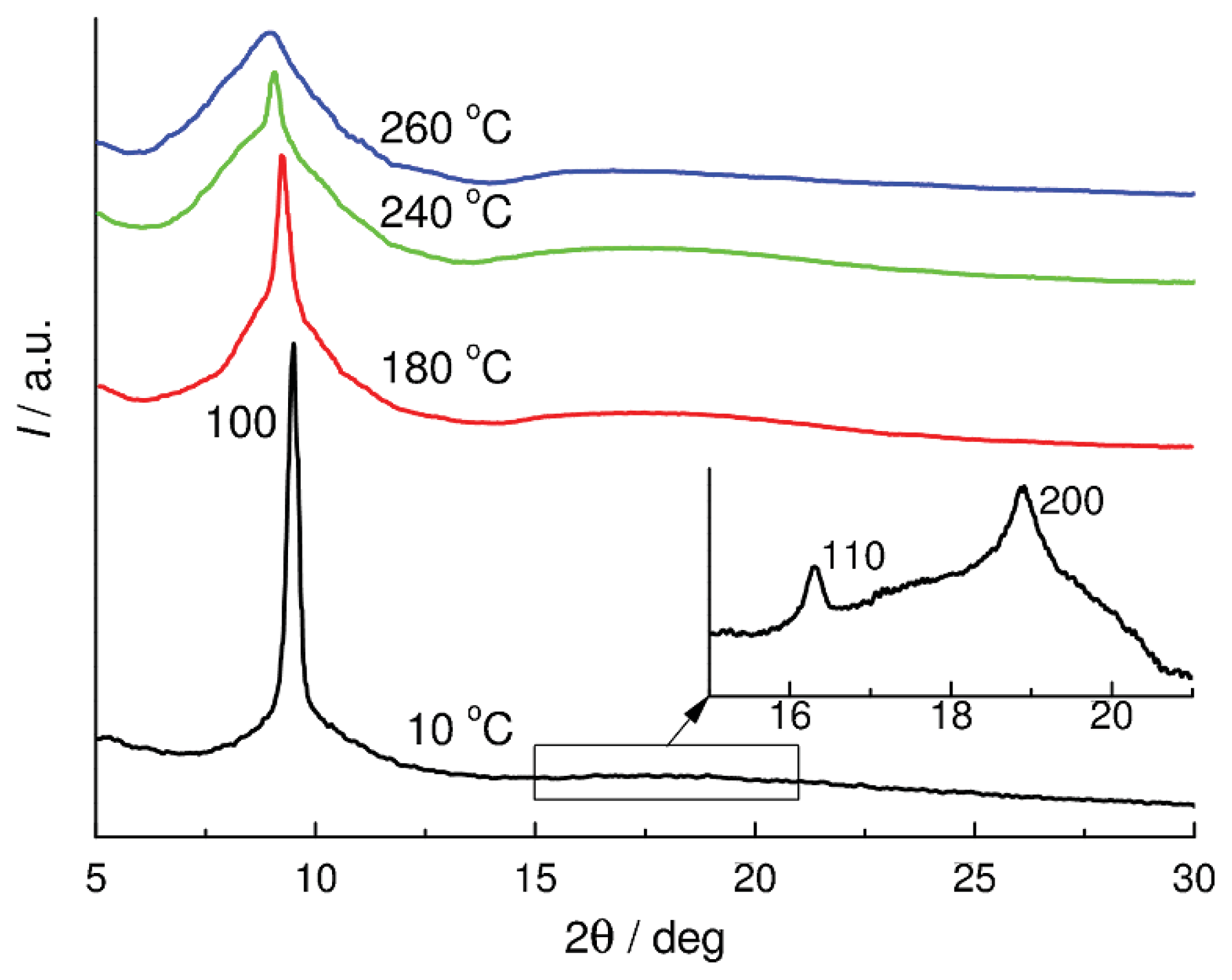
IV-
Et is typical of mesomorphic systems. It exhibits a single narrow reflection in the region 2θ
m = 9.43° (
d1 = 9.37 Å). At low temperatures, two weak reflections were revealed additionally, sin 2θ
m:sin 2θ
I:sin 2θ
II = 1:3:4 (
Figure 1). The aforesaid suggests that in the mesomorphic state, CL POCS
IV-
Et has a 2D packing type with hexagonal symmetry up to
Ti. This packing type in the mesophase is confirmed by the high gradient of the
dm(
T) dependence. The structure-forming element of the mesophase is the macromolecule.
Thus, replacement of the oxygen atom by the (CH2)2 unit between the cyclotetra- or cyclohexasiloxane moieties in the polymer chain leads to the loss of the ability of CL POCS to self-organize with the formation of 1D packing in the case of CL polymethylcarbosiloxanes (PMCS). Additional factors, such as the introduction of ethyl substituents into CL POS and CL POCS, produce intermolecular interactions favoring the recovery of the ability to form the mesomorphic state. This is accompanied by a decrease in the effect of both cyclosiloxane conformations and the chemical structure of the bridge between the cyclosiloxane moieties on Ti.
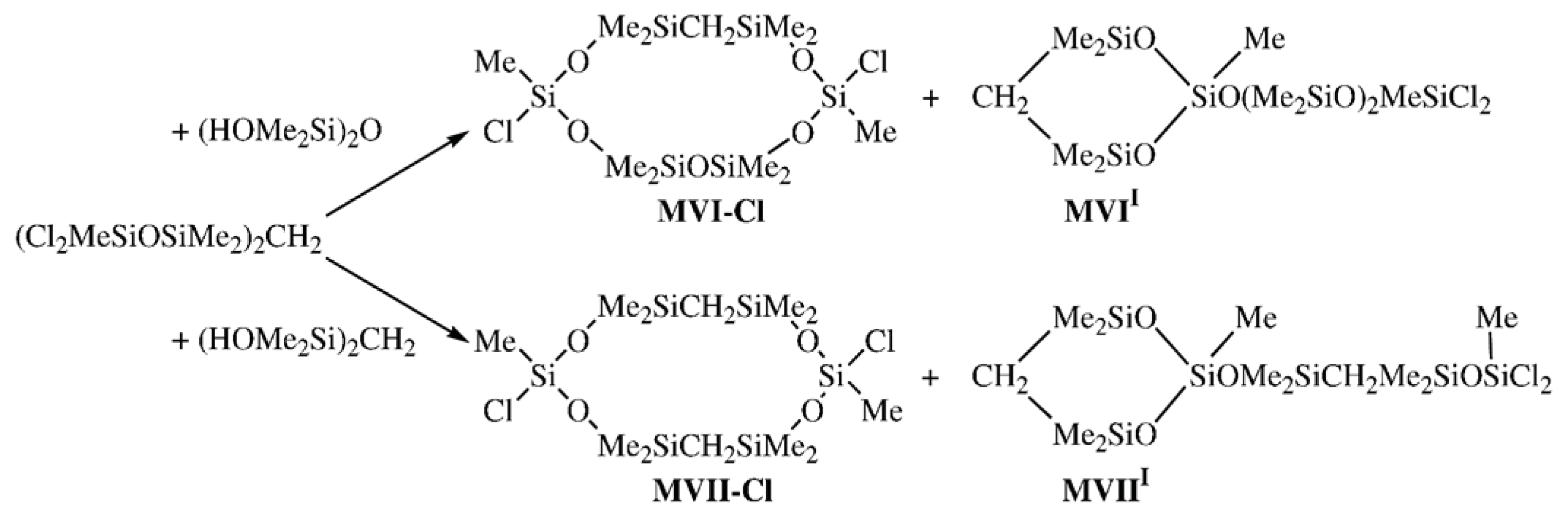
Single oxygen atoms in decamethylcyclohexasiloxane were replaced by CH
2 units by the stepwise condensation of 1,3-dihydroxytetramethyldisilylmethylene with methyltrichlorosilane followed by the interaction of 1,1,7,7-tetrachloro-1,3,3,5,5,7-hexamethyl-4-carbodisiloxane with various organosilicon 1,3-dihydroxy derivatives following
Scheme 2 [
18].
Since our attempt to separate two structural isomers,
MVII-
Cl and
MVIII, had failed, hydrolysis of a mixture of these isomers gave a mixture of hydroxy derivatives; 2,8-dihydroxy-2,4,4,6,6,8,10,10,12,12-decamethyl-5,11-dicarbacyclohexasiloxane (
MVII-
OH) was isolated by fractional recrystallization. To reveal specific features associated with the conformational changes in the cyclohexasiloxane moiety upon the introduction of one and two methylene units, two model monomeric compounds,
viz., 2,8-dihydroxy-2,4,4,6,6,8,10,10,12, 12-decamethyl-5-carbacyclohexasiloxane (
MVI-
OH) and 2,8-dihydroxy-2,4,4,6,6,8,10,10,12, 12-decamethyl-5,11-dicarbacyclohexasiloxane (
MVII-
OH), were studied [
19,
20] by X-ray analysis assuming that the unit of CL POCS would inherit the carbocyclosiloxane conformation. In addition, Compounds
MVI-
OH and
MVII-
OH were used to compare their behaviors on the surface of water under lateral compression in order to evaluate the co-operative contribution of the Me
2SiOSiMe
2 and MeSi(OH)O fragments to the surface of water.
It was found that replacement of an oxygen atom by a CH
2 unit in
trans-2,8-dihydroxy-2,4,4,6,6,8,10,10,12,12-decamethylcyclohexasiloxane (
MII-
OH) lead to small conformational changes (
Figure 2). The angle ∠ SiCSi = 122.1° in the ring noticeably deviated from the tetrahedral angle [
14], whereas the angle ∠ SiCSi in 1,3-disilacyclobutane compounds was close to 90° [
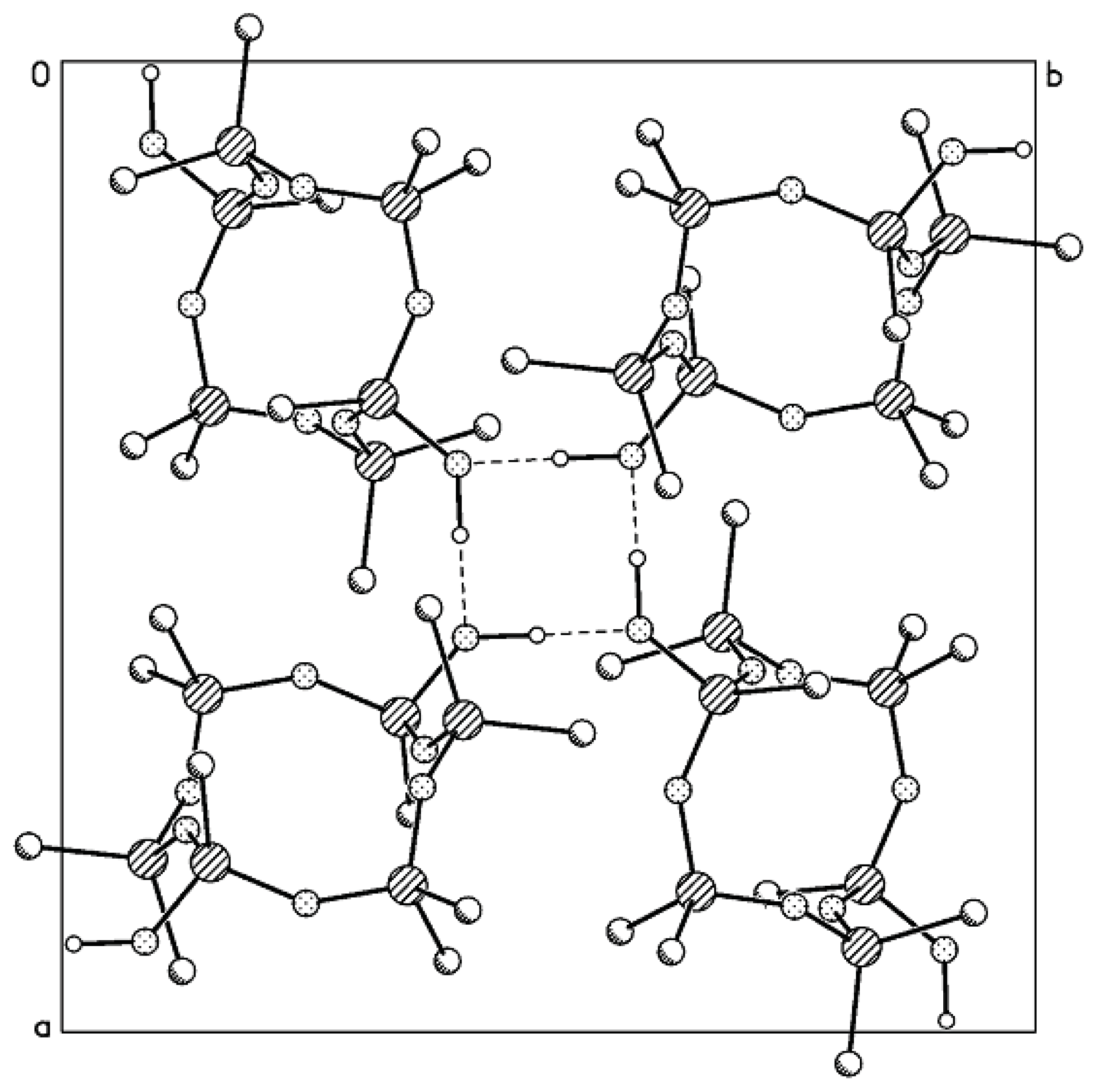
21]. Based on the results obtained, one can state with certainty that the angls ∠ SiCSi in the cyclosiloxane systems can vary within a wide range. In the crystal, four-membered rings comprised of H-bonded
trans-2,8-dihydroxy-2,4,4,6,6,8,10,10,12,12-decamethyl-5-carbacyclohexasiloxane molecules form puckered layers.
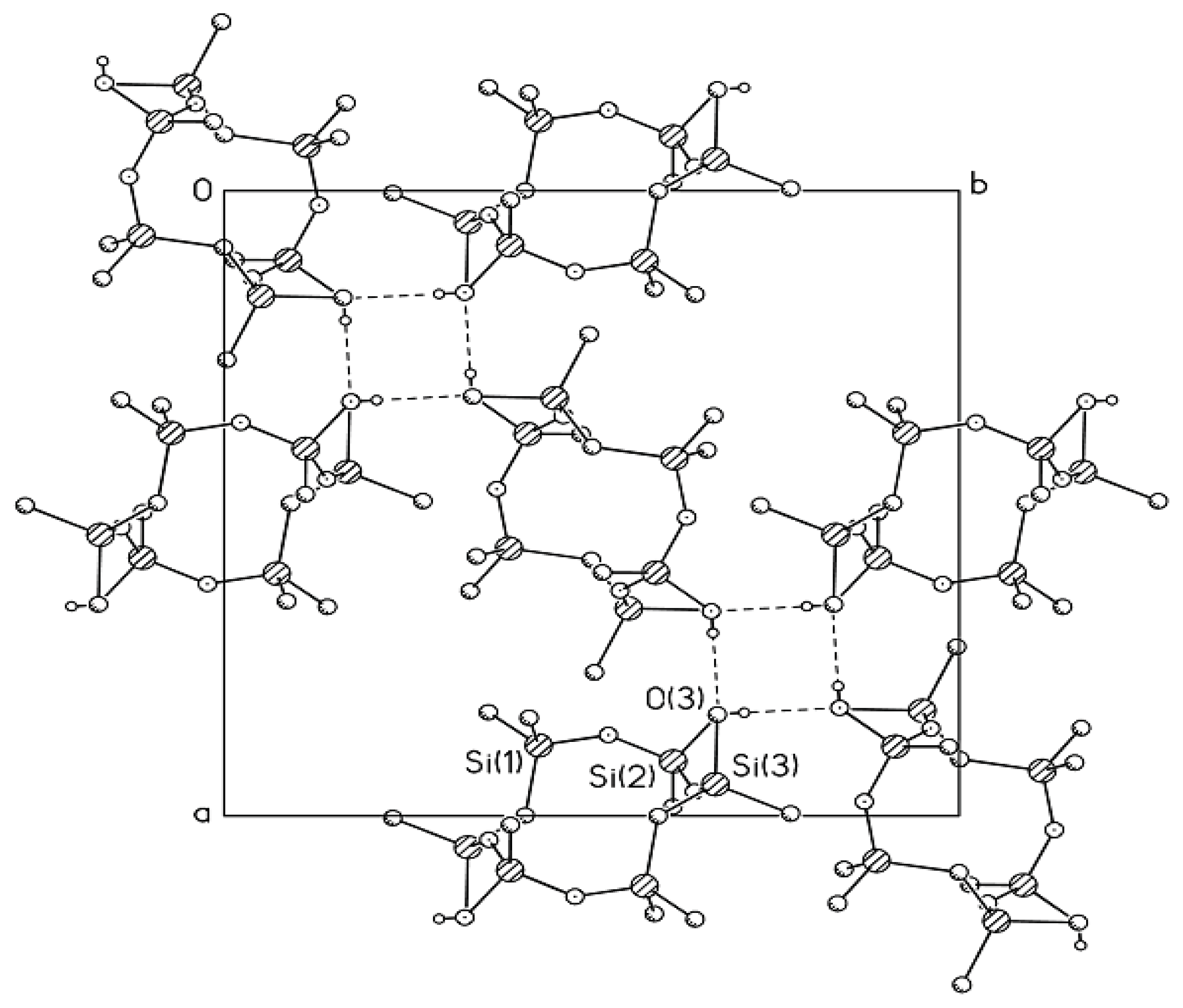
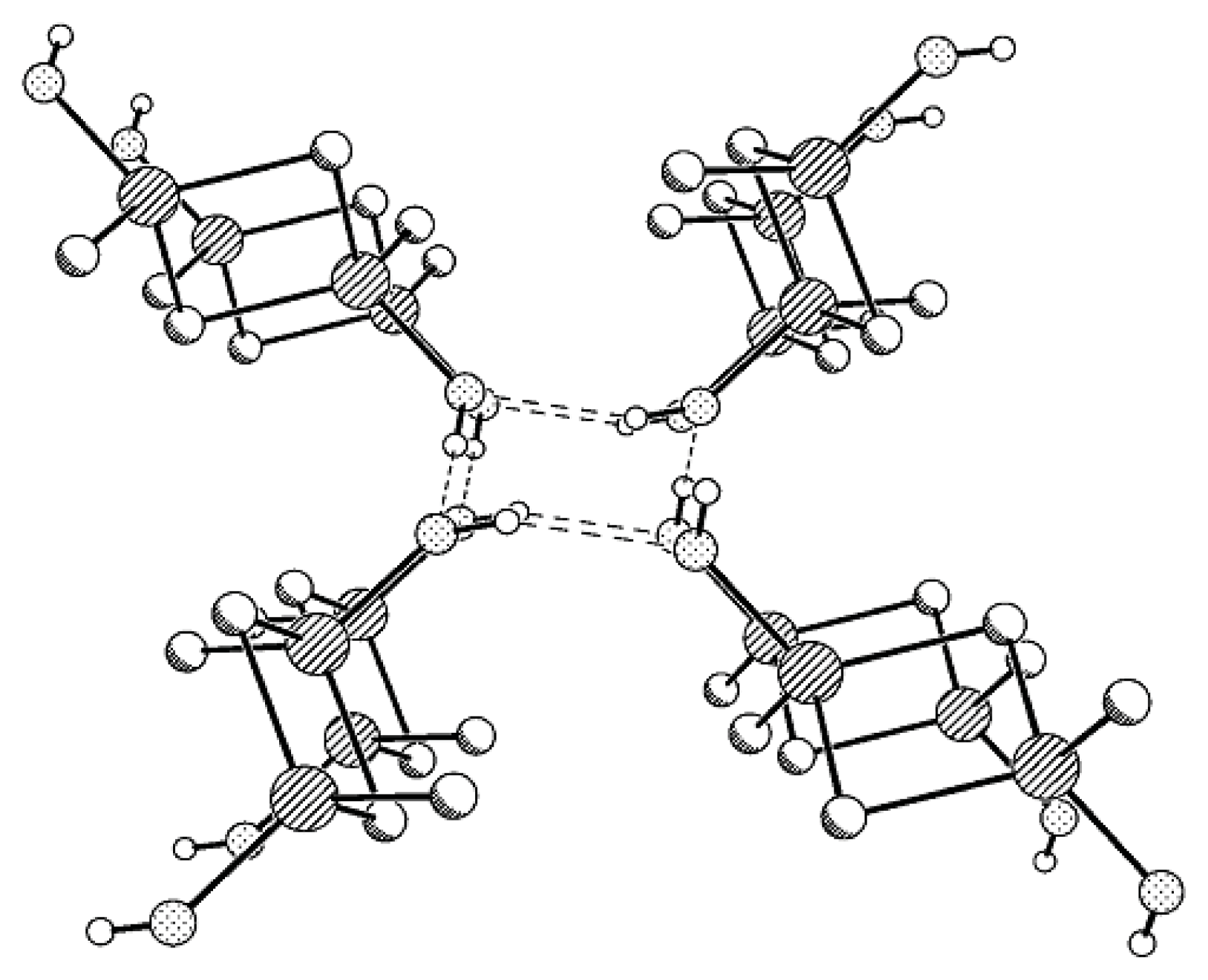
Generally, the conformation of Compound
MVII-
OH with two CH
2 units can be represented by a crown, but in most cases these moieties adopt a complex folded conformation which can hardly be described using some conventional pattern. The valence angle ∠ SiCSi = 123.0° significantly differs from the tetrahedral angle, being, however, close to the angle ∠ SiCSi = 122.1° in Compound
MVI-
OH with one CH
2 unit (
cf. a value of 146.4° for the valence angle ∠ SiOSi in the
trans-Compound
MII-
OH [
22]). The packing of molecules in the crystal of
trans-Compound
MVII-
OH (
Figure 3) is similar to that in the crystal of
MVI-
OH. The molecules are also linked by H-bonds to form tetramers surrounding a fourfold inversion axis. A similar kind of molecular ordering was also found for the
trans-Compound
MII-
OH [
22]. The same space groups, close values of geometric parameters, and similarity in the molecular packing for these three compounds indicate the efficiency of this type of packing methylcyclohexasiloxane molecules. However, an increase in the number of the monomer units with changed conformations in the polymer chain built of such units should be accompanied by changes in their physicochemical properties. In particular, the properties of the resulting polymers can be controlled by choosing the conformational features of monomers taking into account the hydrophilic-hydrophobic nature of the O atoms and CH
2 groups.
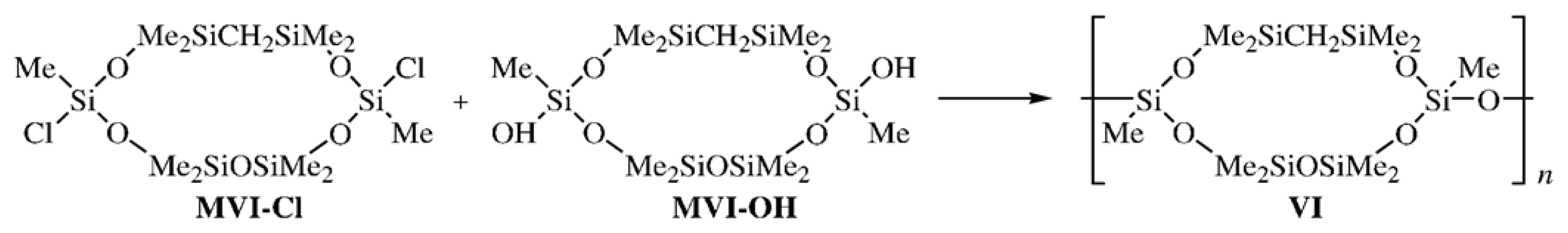
Poly[oxy(2,4,4,6,6,8,10,10,12,12-decamethyl-5-carbacyclohexasiloxane-2,8-diyl)] (
VI) bearing a CH
2 unit in the cyclohexasiloxane moiety was synthesized by the heterofunctional polycondensation of Compound
MVI-
Cl with the
trans-isomer of Compound
MVI-
OH. The structure of CL PMCS
VI was confirmed by
1H and
29Si NMR spectroscopies (
Scheme 3) [
23]:
Differential scanning calorimetry, X-ray, and optical polarization microscopy studies revealed that CL PMCS
VI still could form the mesophase and that the ability to self-organize appeared at a polymerization degree of
n = 14 (
Table 2). Earlier, the ability to self-organize with the formation of a 1D mesophase at a polymerization degree of
n = 3–5 had been reported for CL POS [
14]. As the molecular weight of CL POCS
VI increased to
Mw = 2.55 × 10
4,
Ti increased to 240 °C (
Table 2).
As in the case of CL PMS II-Me, the X-ray diffraction pattern of CL PMCS VI exhibited a strong, narrow reflection at dm = 8.4 Å. This suggests a 1D structural organization of the mesomorphic phase in bulk CL PMCS with the Type-VI structure.
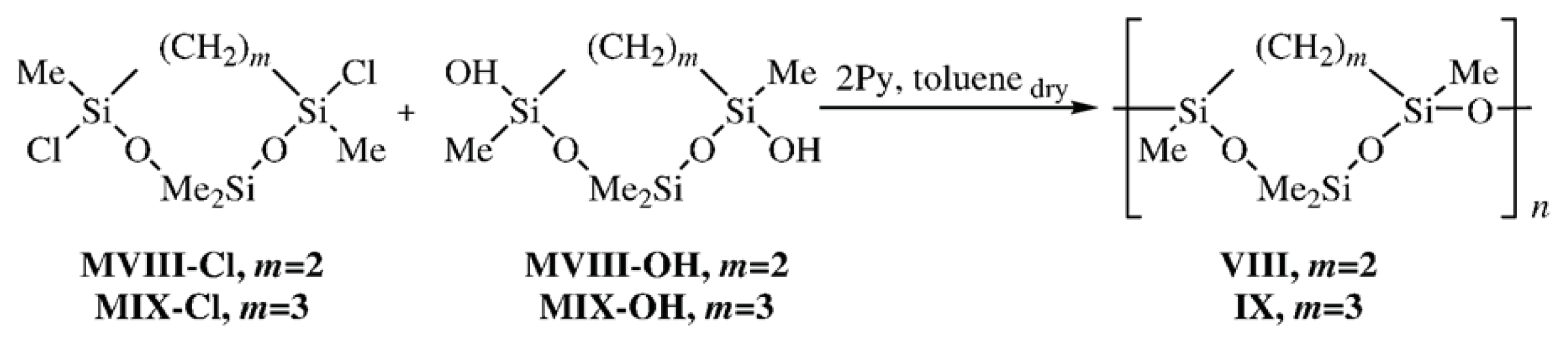
The effect of replacement of an OMe
2SiO unit rather than single O atom by alkylene (ethylene, trimethylene) groups in the cyclosiloxane moiety of the CL PMCS unit on the ability to form the mesophase through the change in the SiO/SiCH
2 ratio in the CL PMCS unit was studied taking oligo[oxy(2,2,4,7-tetramethyl-1,3-dioxa-2,4,7-trisilacycloheptane-4,7-diyl)] (
VIII) and poly[oxy(2,2,4, 8-tetramethyl-1,3-dioxa-2,4,8-trisilacyclooctane-4,8-diyl)] (
IX) as examples. Cyclolinear PMCS
VIII and
IX were obtained by the heterofunctional polycondensation of 4,7-dichloro-2,2,4,7-tetramethyl- 1,3-dioxa-2,4,7-trisilacycloheptane (
MVIII-
Cl) and 4,8-dichloro-2,2,4,8-tetramethyl-1,3-dioxa-2,4,8- trisilacyclooctane (
MIX-
Cl) with the corresponding dihydroxy derivatives
MVIII-
OH,
MIX-
OH following
Scheme 4 [
23]:
The structures of CL PMCS VIII (m = 2) and IX (m = 3) were confirmed by 1H and 29Si NMR data. The 29Si NMR spectrum of Polymer VIII exhibited two groups of signals, viz., a doublet at δ −15.42 and −15.67 for Me2SiO and a triplet at δ −18.99, −19.08, and −19.17 for the MeSi(CH2)2SiMeO fragment of Polymer VIII. The appearance of three signals in a higher field was due to the possibility of three types of joining the units in the polymer chain, namely, cis-trans, trans-trans, and trans-cis. There was no cis-cis combination of units because heterofunctional polycondensation was conducted using the trans-dihydroxy derivative of Compound MVIII-OH. The 29Si NMR spectrum of Polymer IX, similarly to that of CL PMCS VIII, exhibited two groups of signals shifted to a higher field, namely, a doublet at δ −17.55, and −17.76 for Me2SiO, and a triplet at δ −19.91, −19.95, and −19.99 for MeSi(CH2)2SiMeO.
The properties of Polymers
VIII and
IX are summarized in
Table 3. DSC and X-ray studies on the physicochemical properties of Polymers
VIII and
IX showed that the introduction of alkylene groups into the cyclosiloxane moieties of the CL polymer units lead to the formation of amorphous polymers in the temperature range from −140 to 200 °C despite the fact that Polymers
VIII and
IX were enriched with
trans-
trans junctions. Polymers
VIII and
IX are characterized by the same
Tg. The lack of the mesomorphic state in these polymers is most likely due to the asymmetric structure of the silaoxacycloalkane moiety and atactic structure of the polymer chain.
To compare the physicochemical properties of CL PMCS with asymmetric (
VIII and
IX) and symmetric methyldisilaoxacycloalkane moieties, methods of synthesis of bifunctional compounds 1,3-dimethyl-1,3-disilacyclobutane and 1,4-dimethyl-1,4-disilacyclohexane [
24,
25] were developed. Cyclolinear poly[oxy(1,3-dimethyl-1,3-disilacyclobutane-1,3-diyl)] (
X) was synthesized by the heterofunctional polycondensation of 1,3-dichloro-1,3-dimethyl-1,3-disilacyclobutane (
MX-
Cl) with 1,3-dihydroxy-1,3-dimethyl-1,3-disilacyclobutane (
MX-
OH) following
Scheme 5 and poly[oxy(1,4-dimethyl-1,4-disilacyclohexane-1,4-diyl)] (
XI) was obtained in the reaction of 1,4-dichloro-1,4-dimethyl-1,4-disilacyclohexane (
MXI-
Cl) with 1,4-dihydroxy-1,4-dimethyl-1, 4-disilacyclohexane (
MXI-
OH) following
Scheme 6:
Cyclolinear PMCS X and XI are attractive models for comparing the effects of the ratio of the number of polar to that of nonpolar groups (SiO and SiCH2, respectively) and the length of hydrophobic fragments in the unit of CL polymers on the intermolecular interactions in the bulk and in a monolayer on the surface of water.
The crystal structures and molecular geometries of Compounds
MX-
OH [
21] and
MXI-
OH [
20] whose conformational features should be retained in the unit of the polymer chain were studied by X-ray analysis.
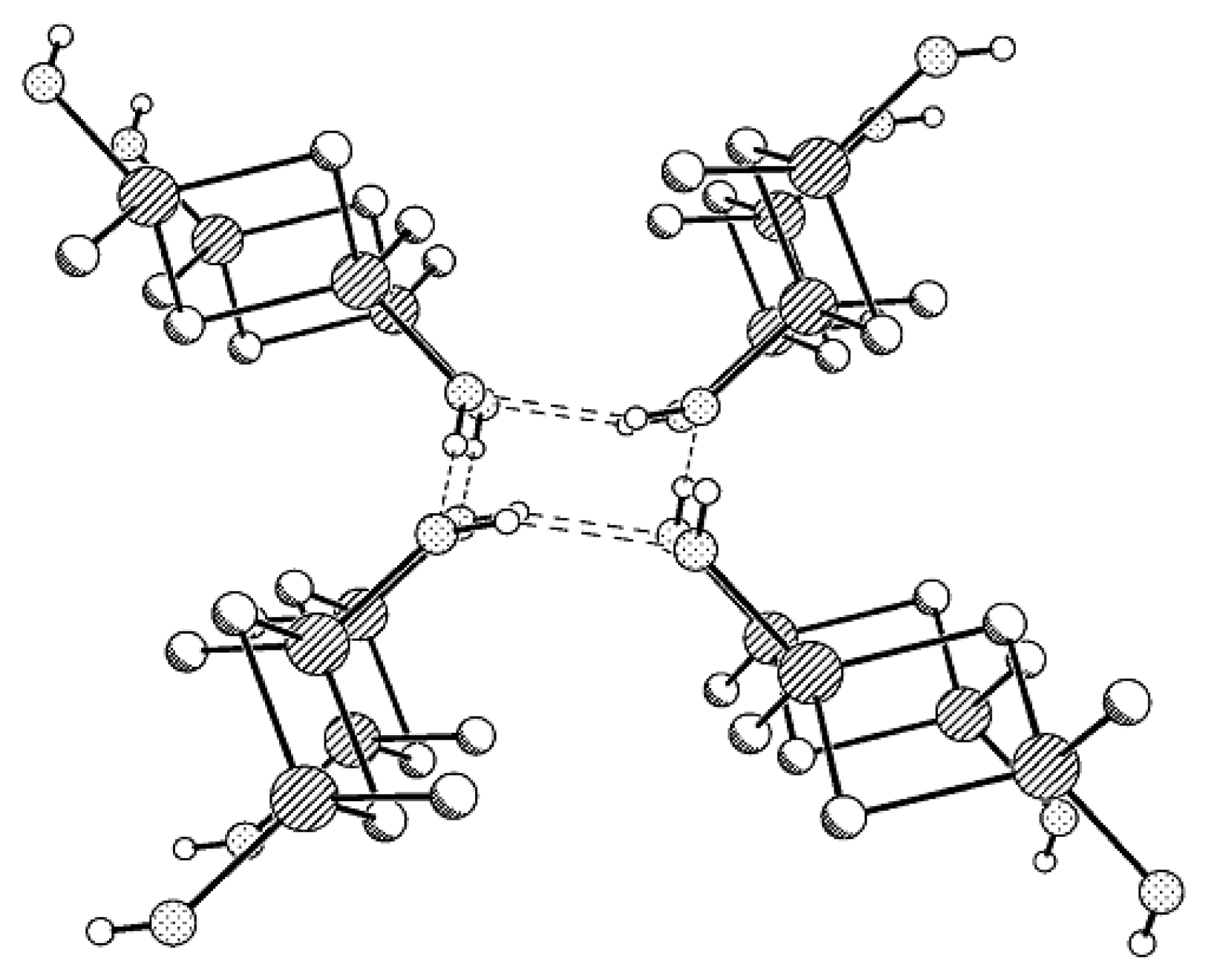
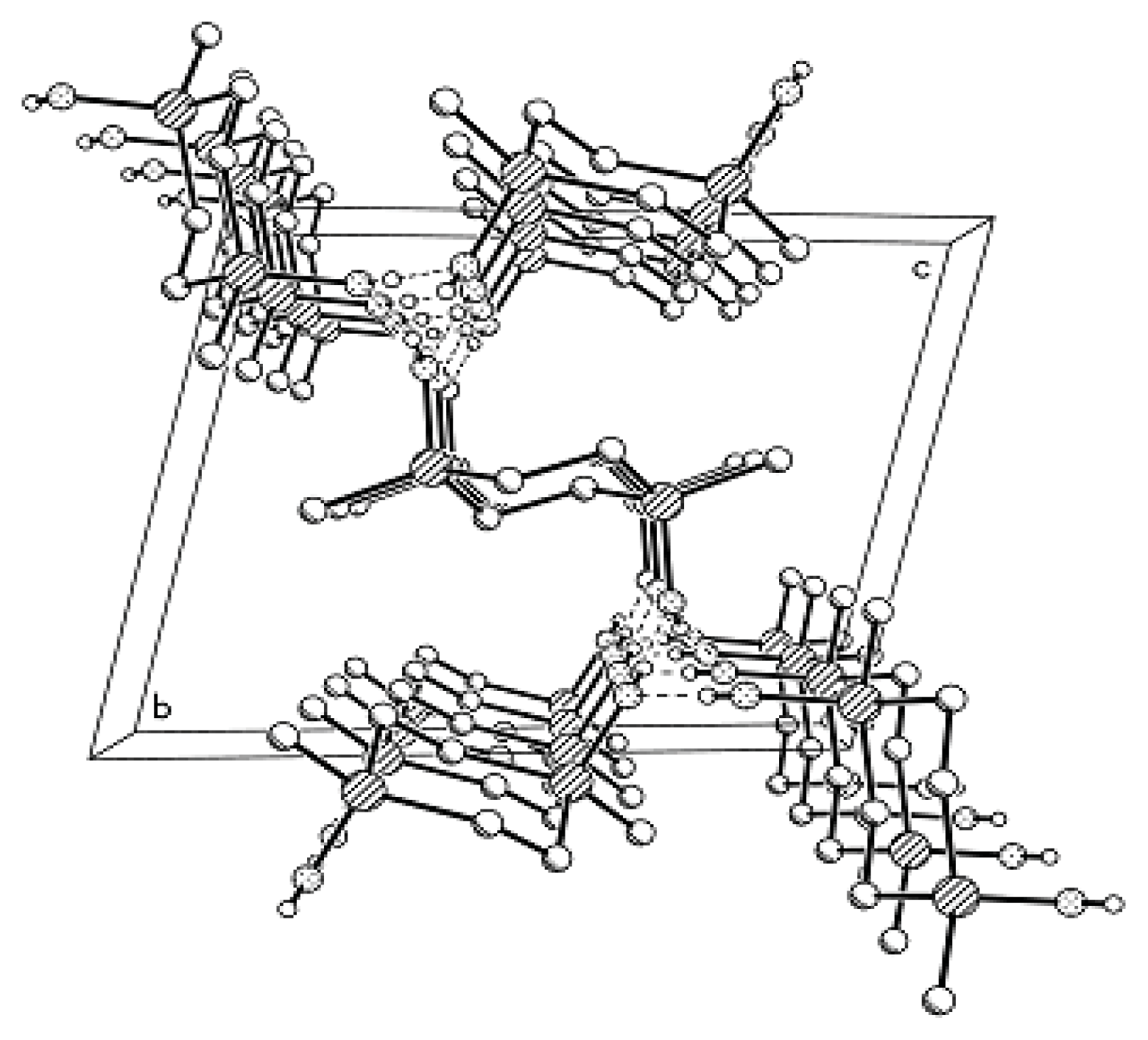
Compound
MX-
OH forms H-bonded associates containing two types of independent molecules. Four
MX-
OH molecules participate in the formation of a four-membered H-bonded ring, but, unlike Compounds
MII-OH,
MVI-OH, and
MVII-OH characterized by layered packing type, in the structure of
MX-OH the plane of one disilacyclobutane moiety makes some angle with the plane of the other disilacyclobutane moiety, and this pattern is repeated along the axis (
Figure 4).
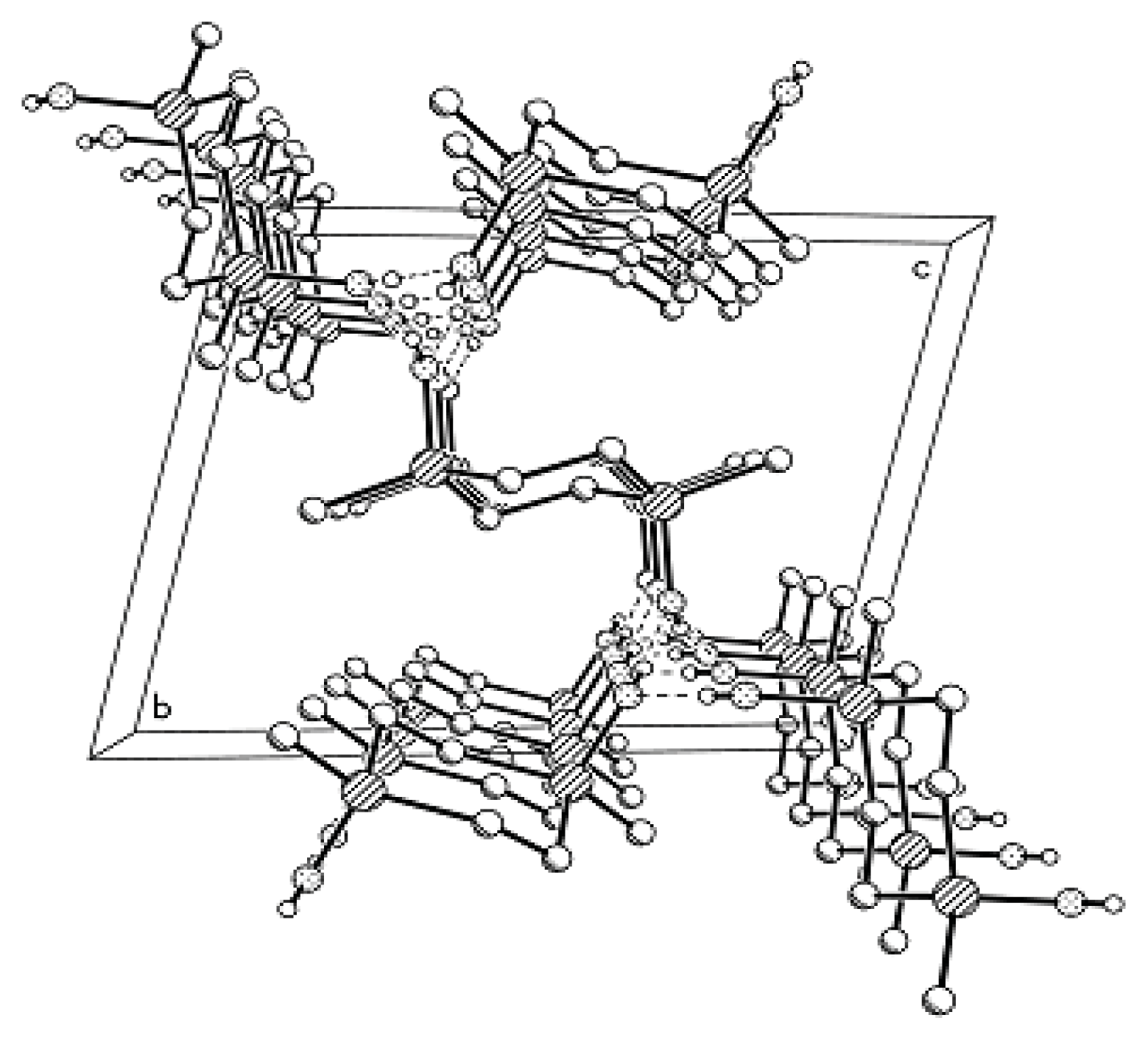
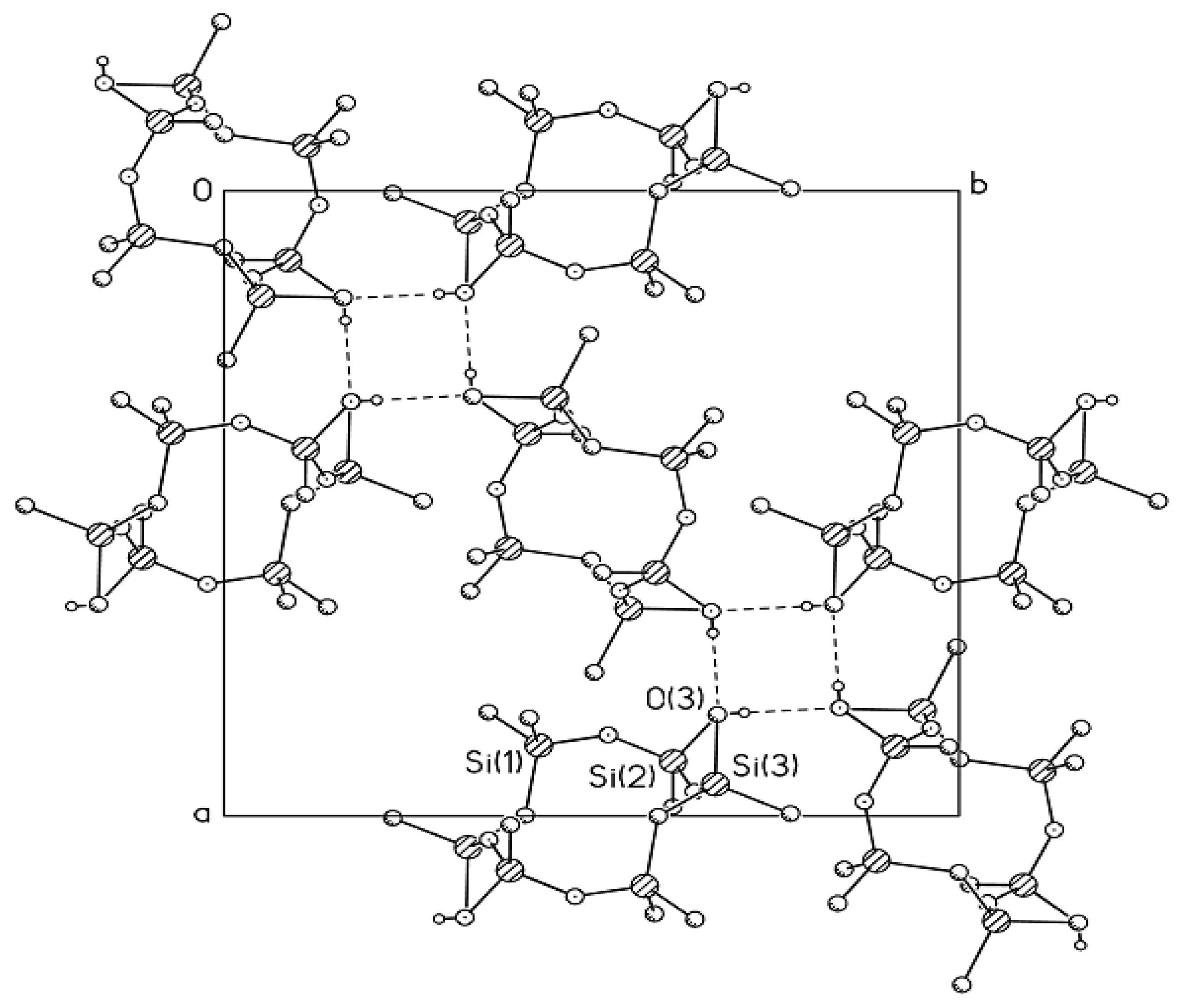
In the crystal structure of Compound
MXI-OH, the unit cell contains three independent molecules with very similar conformations to the disilacyclohexane moieties adopting the chair conformation (
Figure 5). Silicon atoms deviate from the mean plane of carbon atoms. Here, one deals with the classical tetrahedral valence angles at carbon atoms. In the crystal, the
MXI-OH molecules form columns linked by an infinite system of H-bonds along the axis. As in Compound
MX-OH, the centers of three H-bonded
MXI-OH molecules do not lie in the same plane.
Thus, the columnar crystal structures of the trans-dihydroxy derivatives of methyldisilacycloalkanes are fundamentally different from the layered structures of Compounds MII-OH, MVI-OH, and MVII-OH.
X-ray and gel permeation chromatography (GPC) studies showed that the CL PMCS with the Type-X unit structure was a completely soluble, amorphous polymer characterized by a polydispersity index Mw/Mn of 2.57 and Mw = 1.01 × 105.
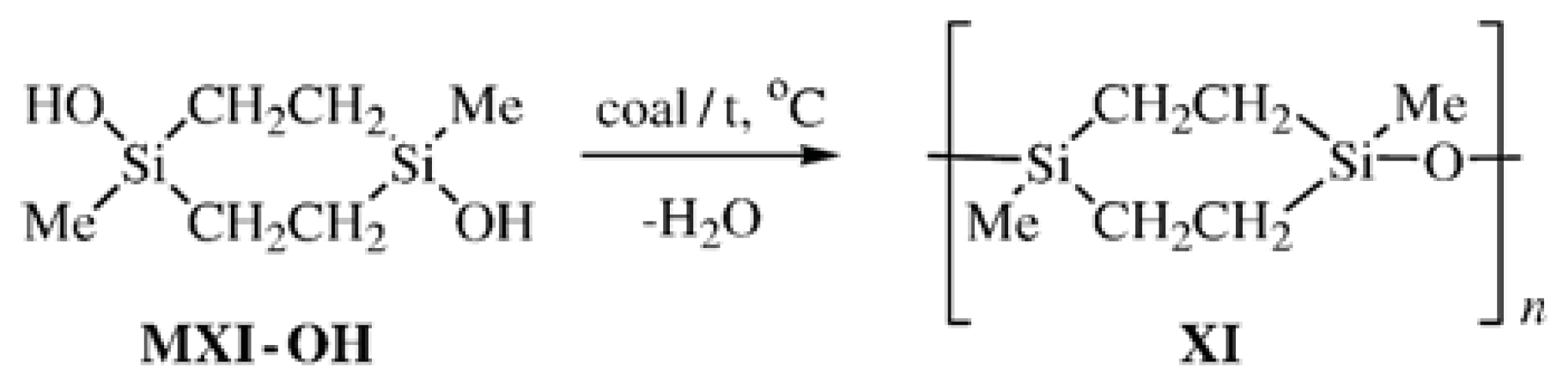
Cyclolinear PMCS
XI was synthesized by the heterofunctional polycondensation following
Scheme 6 and by the homocondensation of a mixture of isomers of Compound
MXI-OH in the presence of activated carbon following
Scheme 7:
The
1H NMR spectrum of Polymer
XI exhibits a singlet from CH
3 protons in the region δ 0.03 or a triplet at δ 0.024, 0.028, 0.030 and a multiplet from CH
2 protons in the region δ 0.74–1.28. The
29Si NMR spectrum of Polymer
XI demonstrates three multiplets with the same Δδ values (0.05–0.07 ppm) instead of a triplet or quartet with different signal intensity ratios. The multiplet pattern of the
29Si NMR spectrum of CL PMCS
XI reflects the variety of inequivalent silicon atoms in the polymer backbone. Each group of signals corresponds to different combinations of cyclohexane moieties in the polymer chain and characterizes the presence of three conformers with axial, equatorial, and axial-equatorial arrangement of substituents in the chain. The most probable conformation of the polymer chain of CL PMCS
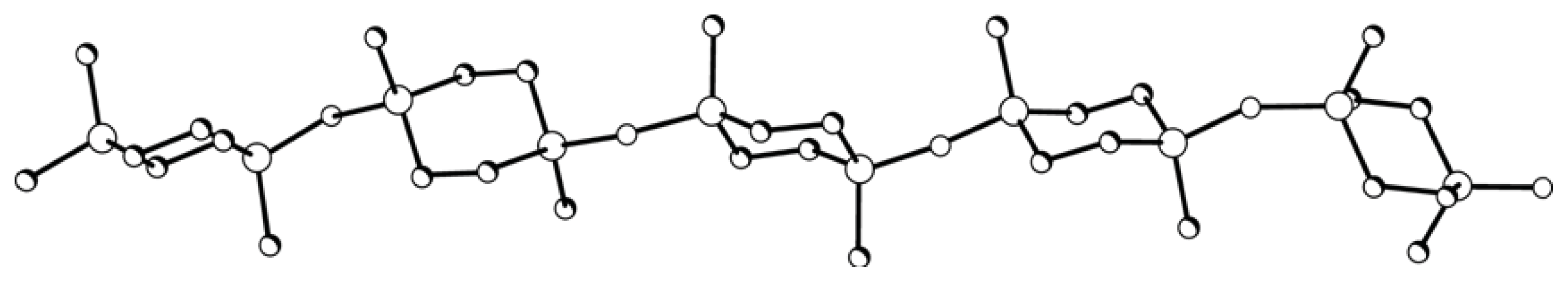
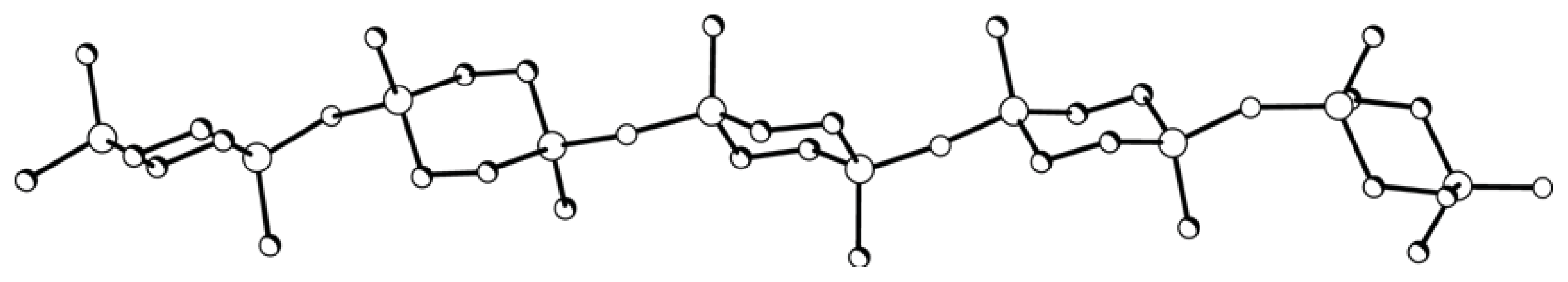
XI was calculated by the molecular mechanics method. A chain segment of Polymer
XI comprising five monomer units shows no translational ordering of disilacyclohexane moieties relative to one another along the chain (
Figure 6).
Based on the NMR spectroscopy, infrared (IR) spectroscopy, and GPC data, the CL PMCS
XI with the most regular structure was obtained by the heterofunctional polycondensation (
Scheme 6). X-ray data for the molecules of Compound
MXI-OH in the crystal [
20] suggest another type of association compared to that found for Compounds
MII-OH,
MVI-OH, and
MVII-OH. Compound
MXI-OH goes into the mesomorphic state upon melting [
25]. The X-ray diffraction pattern of Compound
MXI-OH obtained at 132 °C shows no reflections from the crystal and only a narrow reflection at 2θ = 8.86° (
d1 = 9.97 Å) remains. The DSC trace of Compound
MXI-OH exhibits two endothermic peaks in the temperature range of 124–160 °C. The peaks are shifted toward lower temperatures on re-heating. Unlike Compound
MXI-OH, the DSC traces of CL PMCS
XI show
Tg = −54 to −55 °C irrespective of the method of synthesis, and only the syndiotactic Polymer
XI obtained from pure
trans-isomers of the monomers demonstrates an endothermic transition with Δ
H = 1.1 J g
−1 in the temperature range of 30–58 °C.
2.2. The Bulk Behavior of CL POS and CL PMCS
Tables 1,
2 and
3 present the properties of CL Polymers
I–
XI with different numbers of SiO and SiCH
2 groups in the unit. As can be seen that the molecular weights of all CL polymers are not high and that the degree of polymerization varies from 14 to 175. For some CL polymers (e.g.,
II and
VI), different molecular weights are given, and it is shown that the
Ti values of the polymers increase with increasing the molecular weight. Polymers
II and
VI with different numbers of SiO and SiCH
2 groups are characterized by close
Ti values. The ability of CL PMS
VI to form the mesomorphic phase appears at much lower degrees of polymerization compared to linear polydiorganosiloxanes, e.g., polydiethylsiloxane and polydi-
n-propylsiloxane [
15,
16].
A comparison of the Tg and Ti values for the CL POS I-Me and I-Et and CL POCS III-Me and III-Et shows that Tg considerably increases upon replacement of the oxygen atom by the CH2CH2 unit due to a higher flexibility of the >SiMeOMeSi< group compared to the >SiMeCH2CH2MeSi< fragment. The CL POCS bearing methyl substituents are prone to crystallization. Unlike CL POS I-Me, Polymer III-Me does not undergo a transition to the mesomorphic state upon melting. If the introduction of ethyl substituents into the CL POS I-Et causes an abrupt extension of the existence region of the mesophase, Tg of CL POCS III-Et is reduced by more than 40 °C, but the polymer is in the amorphous state unlike I-Et. Thus, the introduction of the CH2CH2 bridge between the cyclotetrasiloxane moieties causes changes in the mutual arrangement of the two ring planes at a fixed angle ∠ SiCH2CH2, whereas in the case of CL POS I-Me, I-Et, and I-Pr, the angle ∠ SiOSi can vary between 140° and 180° depending on the size of the substituents; this plays the decisive role in the formation of the 1D or 2D type of packing in CL organosilicon polymers.
A comparison of the properties of the CL POS
II-
Me and
II-MePh and those of the CL POCS
IV-
Me,
IV-
Et, and
VI-
Me shows that replacement of the oxygen atom in the bridge by a CH
2CH
2 unit leads to the loss of the ability to self-organize. At the same time, replacement of one oxygen atom in the cyclohexasiloxane moiety by methylene unit has no effect on the transition temperatures
Tg,
Ti and the existence region of the mesomorphic state; therefore, the mutual position of the two moieties in the chain should be considered as the key factor. Note that for Polymer
VI-
Me the dependence of
Ti on the molecular weight is the same as that for Polymer
II-Me. Thus, at the same SiO/SiCH
2 ratio in the main chain of CL polymethylcarbosiloxanes
IV-Me and
VI-Me, the ability to self-organize in bulk is lost for CL PMCS
IV-Me. However, Compounds
MII-OH and
MVI-OH in the crystalline phase adopt a crown conformation and form a layered H-bonded packing.
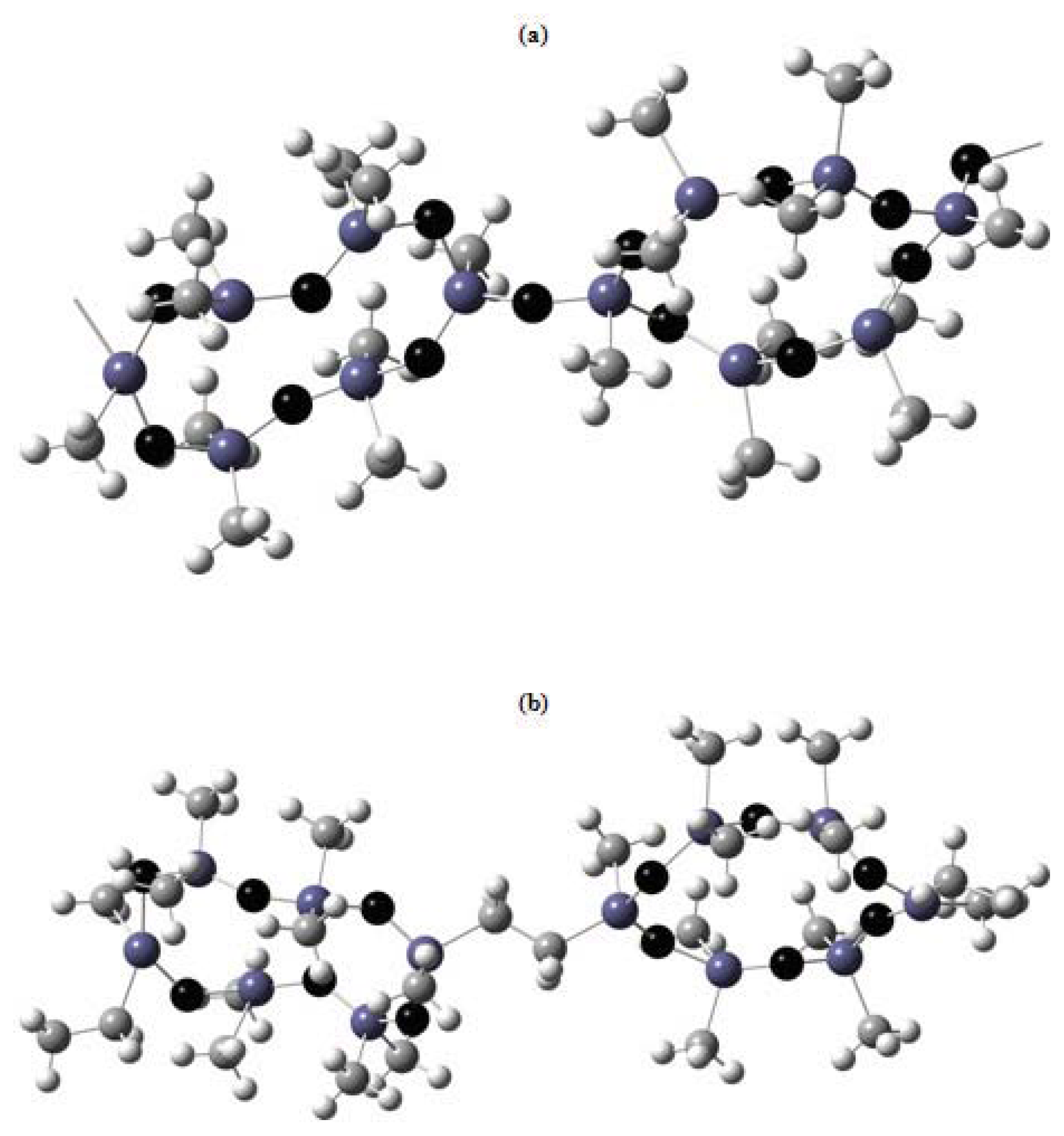
Figure 7 presents the possible types of joining in the CL PMS and CL PMCS backbones. From the data for CL PMS and CL PMCS it follows that replacement of the oxygen bridge between the rings by CH
2CH
2 leads to a change in the type of macromolecular packing.
Table 3 lists the properties of CL PMCS
VIII and
IX with ethylene and trimethylene groups, respectively, in the trisilaoxacycloalkane moieties. The data in
Table 3 show that replacement of the OMe
2SiO group in CL PMS
I-Me by aliphatic groups causes
Tg to increase irrespective of the chain length of the (CH
2)
n fragment. However, the
Tg values for Polymers
VIII and
IX remain unchanged; above
Tg, the polymers are amorphous. A decrease in the number of organic substituents at silicon atom in the methylsilaoxaalkane units leads to weakening of intermolecular interactions. In addition to the Polymers
VIII and
IX with asymmetric structures of the chain units, Polymers
X and
XI with symmetric structures of the units were obtained. Their properties are also listed in
Table 3. Despite the fact that the SiO/SiCH
2 ratio in the unit considerably decreases (SiO/SiCH
2 = 0.50 for
X and 0.25 for
XI),
Tg changes insignificantly compared to the
Tg values of Polymers
VIII and
IX. Above
Tg, CL PMCS
XI is in the mesomorphic state, as confirmed by the DSC and optical polarization microscopy data. However,
Ti of Polymer
XI is much lower than those of the CL PMS
I-Me,
II-Me or
VI-Me, although some increase in
Ti of Polymer
XI with an increase in its molecular weight is quite probable. Nevertheless, the degree of polymerization (
n) of Polymer
XI is
n = 30, being comparable with the
n values for the two Polymers
VI (
Table 2). A comparison of these data suggests a strong effect of diphilic character of the monomer unit of CL Polymers
I-Me,
II-Me, and
VI-Me on the intermolecular interactions and conservation of 1D ordering in a wide temperature range in these systems.
The aforementioned results are related to self-organization of cyclolinear organosilicon polymers in bulk. Recently, the data on self-organization of
trans-tactic CL organosiloxane copolymers with different structures of the monomer unit were published [
26,
27]. In the work [
26] the ability of coexistence of two types of macromolecular packing of poly[oxy(2,4,4,6,6,8,10,10,12, 12-decamethylcyclohexasiloxane-2,8-diyl)-dimethylsiloxane] in crystalline and mesomorphic states was demonstrated using different methods. Later, the conditions allowing formation of either one or the other packing were found for the
trans-tactic poly[oxy(2,4,4,6,6,8,10,10,12, 12-decamethylcyclohexasiloxane-2,8-diyl)-methylvinylsiloxane]. These data will be presented in the next communication.
2.3. The Behavior of CL POS and CL PMCS at Air/Water Interface
Earlier [
5], it was reported that CL POS could form ordered monolayers. In some cases, destruction of a monolayer is accompanied by the formation of discrete multilayers. The results of the study of the behavior of CL POS on the surface of water show that the chemical structure, namely, the size of the cyclosiloxane moiety, as well as its symmetry and spatial isomerism affects the ability of CL POS to form mono- or multilayers on the surface of water. In addition to these factors, the degree of polymerization and organic substituents at silicon atoms also contribute to the self-organization of CL POS at the air/water interface. A study of the dependence of the surface pressure (π) on the surface area per monomer unit of CL PMS on the size of the cyclosiloxane moiety showed that the collapse surface pressure of the monolayer π was 10 ± 0.5 mN m
−1 for CL PMS with the chain unit from cyclotetra- to cycloheptasiloxane. It is noteworthy that no unambiguous dependence of π on the number of SiOSi groups in the cyclosiloxane unit of CL PMS was observed [
5].
In this work, we studied the influence of changes in the hydrophilic–hydrophobic balance of macromolecules attained by introducing hydrophobic groups into the backbones of two CL POS, namely, poly[oxy(2,4,4,6,8,8-hexamethylcyclotetrasiloxane-2,6-diyl)] (I-Me) and poly[oxy(2,4,4,6,6,8,10,10,12,12-decamethylcyclohexasiloxane-2,8-diyl)] (II-Me), on:
the ability to form a Langmuir monolayer;
the limiting surface pressure of monolayer collapse; and
the ability to self-organize into multilayer structures upon collapse of the Langmuir monolayer.
Earlier [
28], we had studied the effect of hydrophobic groups in 1,3- and 1,4-decamethylcyclohexasilane moieties in CL methylsilanesiloxane copolymers on the surface properties at the air/water interface. It was found that the π value for methylsilanesiloxane copolymers abruptly decreased to 5.0 mN m
−1 (
cf. π ≈ 10 mN m
−1 for CL PMS
I-
Me and
II-
Me). At the same time, it was shown that 1,3-dihydroxy- and 1,4-dihydroxydecamethylcyclohexasiloxanes could spread at the air/water interface with the formation of monolayers.
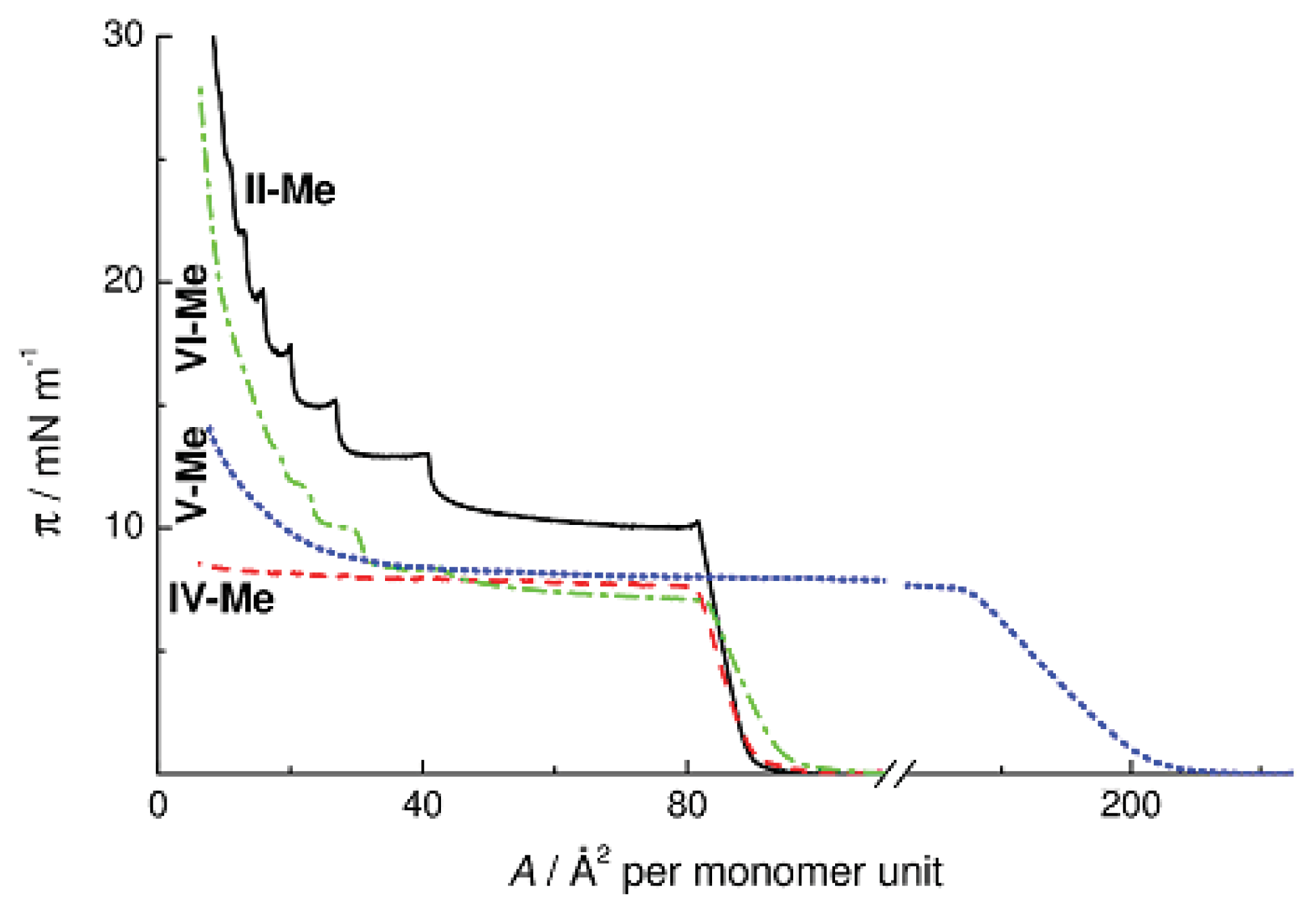
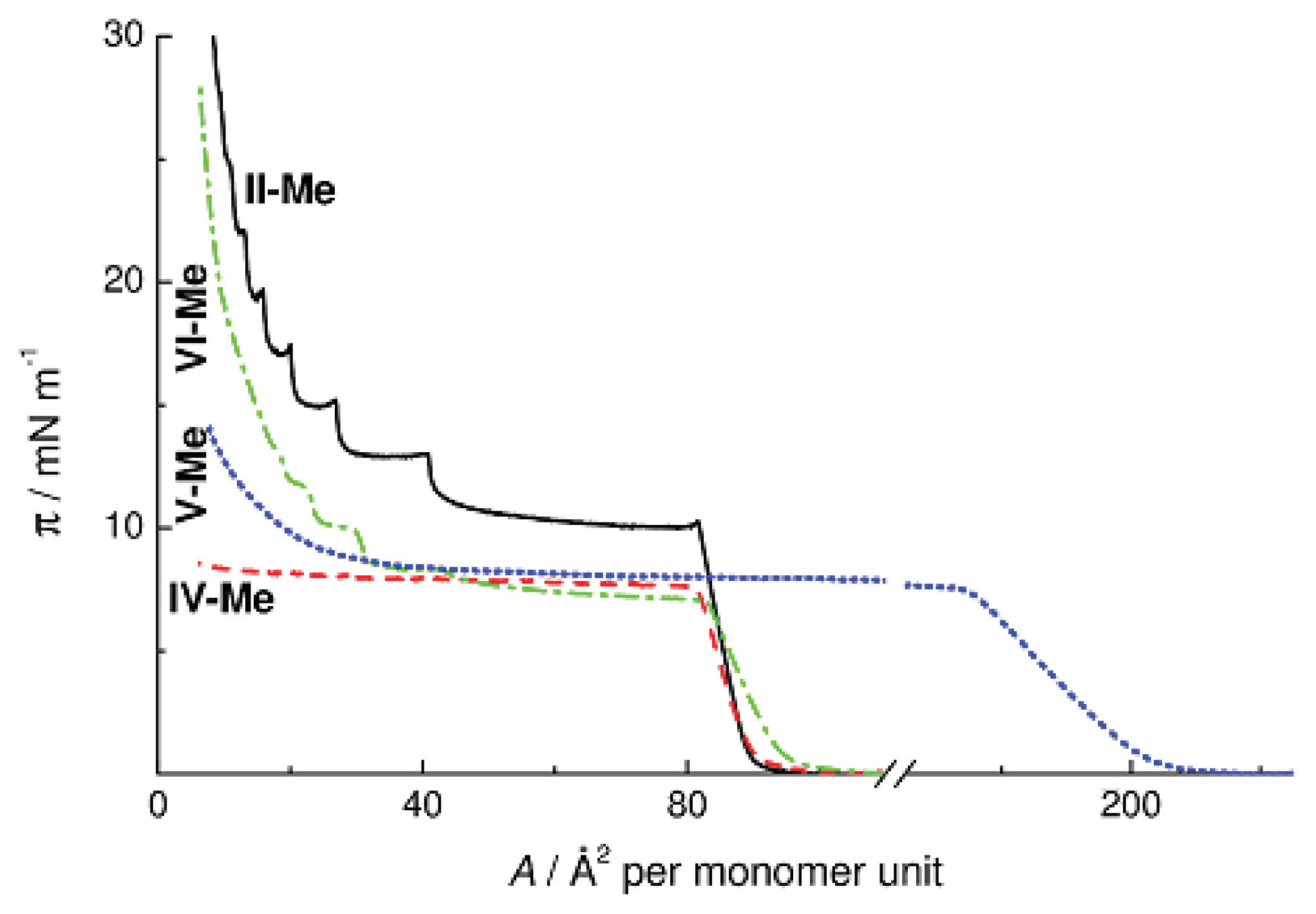
Figure 8 presents the π-
A isotherms of methyl substituted Polymers
II and
IV–
VI with different structures of monomer units. Replacement of an oxygen atom by a hydrocarbon fragment (
IV-Me and
VI-Me) causes no changes in the surface area occupied by the monolayer on the surface of water; at the same time, the surface pressure decreases by 2.5–3 mN m
−1. Thus, macromolecules on the surface are arranged similarly, and the π value decreases because the methylene units are surface inactive. On the other hand, the π-
A isotherms of Polymers
IV and
VI have fundamentally different shapes. Replacement of the oxygen bridge in Polymer
IV-Me by the hydrophobic ethylene group leads to the loss of the multilayer character of monolayer collapse, whereas replacement of the ring oxygen atom by a methylene unit in Polymer
VI causes no changes in it. The character of the surface pressure isotherm of Copolymer
V-Me with ethylene bridges between alternating cyclohexa- and cyclooctasiloxane moieties is similar to the case of CL PMCS
IV-Me. Thus, it is the inter-ring oxygen atom that determines the character of intermolecular packing of polymer chains in the cyclohexasiloxane monolayers. Replacement of this oxygen atom by the ethylene bridge causes suppression of the mesophase in the bulk of the sample as well as the loss of the multilayer character of the π-
A isotherm on the water surface for Polymers
IV-Me and
V-Me.
Our study was in particular aimed at attaining successive replacement of oxygen atoms in the cyclosiloxane moiety by CH
2 units. Unfortunately, the CL PMCS based on the Monomer
MVII-OH was not obtained experimentally because an attempt to separate the starting structural isomers
MVII and
MVIII had failed. In this connection, a comparative assessment was made by studying the behavior of monomeric Compounds
MVI-OH and
MVII-OH on the surface of water.
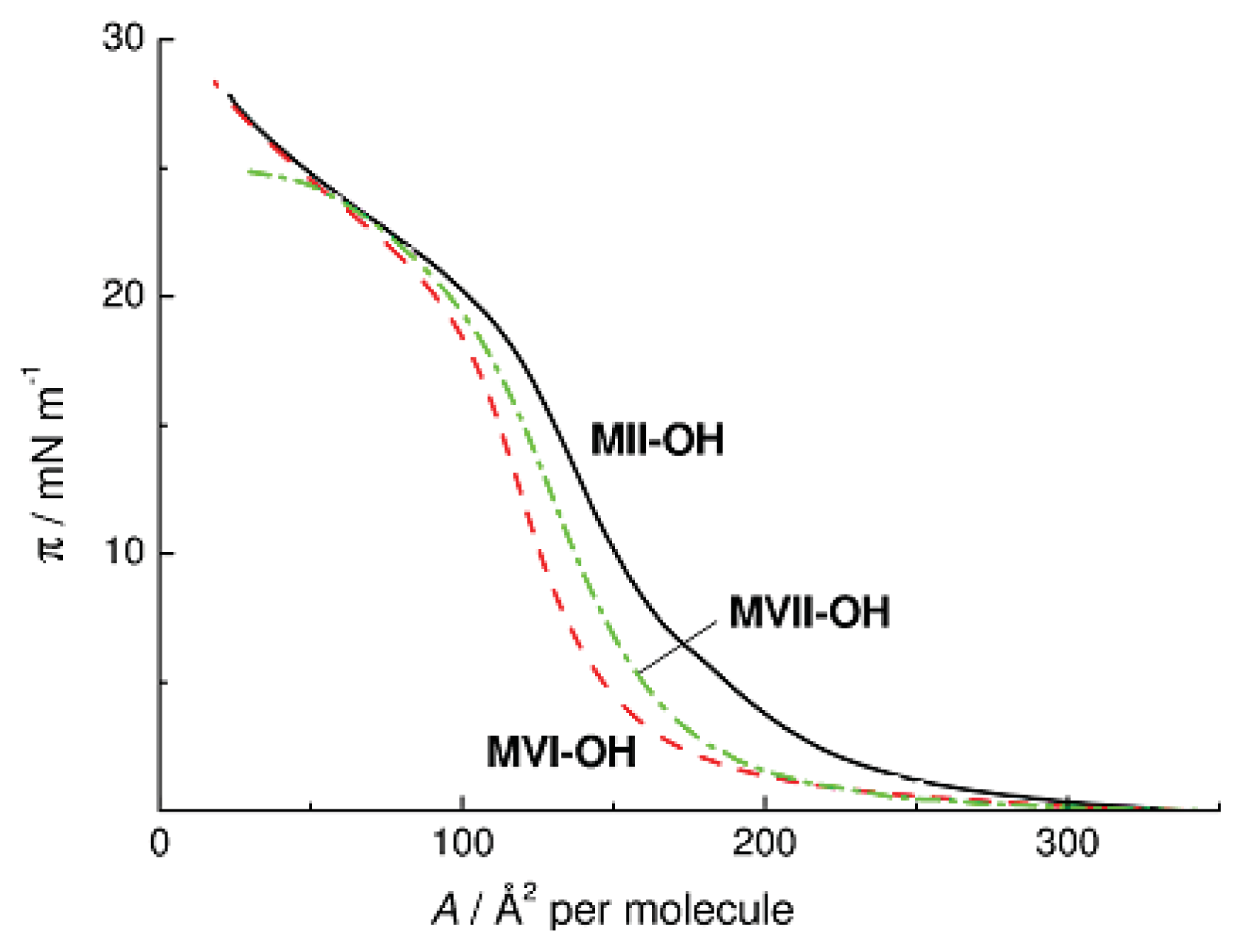
Figure 9 presents the π-
A isotherms of Compounds
MII-OH,
MVI-OH, and
MVII-OH. As can be seen, the three isotherms of the monomeric compounds have similar shapes and close maximum π values. It follows that the major contribution to the interaction with the water surface during association of
MII-OH,
MVI-OH, and
MVII-OH through formation of intermolecular H-bonds comes from OH groups. The SiOSi groups make a less significant contribution to the interaction of cyclosiloxanes with the surface of water.
Replacement of the OMe
2SiO fragment of the monomer unit in CL POS
I-Me by ethylene and trimethylene groups led to Polymers
VIII and
IX, respectively.
Figure 10a presents a two-step π-
A isotherm of Polymer
IX similar to that of the surface pressure isotherm of linear PDMS [
8]. By analogy with PDMS, the second transition can be interpreted either as transition to a helical conformation or as the formation of a bilayer, because the pattern of siloxane bonds in the unit of Polymer
IX is similar to that in linear PDMS and the trimethylene bridge is sufficiently long. The collapse surface pressure of the monolayer of Polymer
IX decreases to 5.8 mN m
−1 (
cf. π = 10.5 mN m
−1 for CL PMS
I-Me).
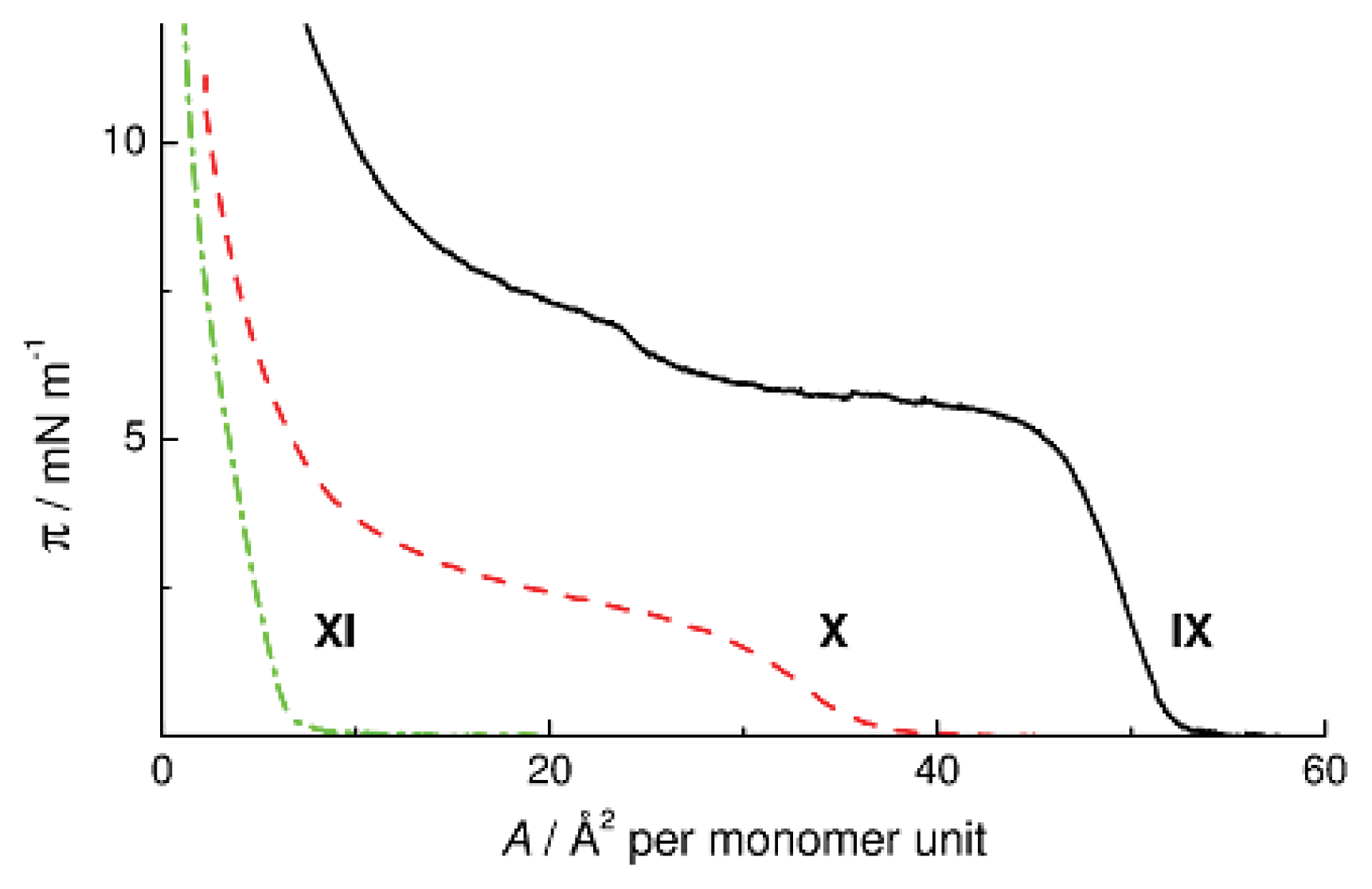
Cyclolinear PMCS
X and
XI are attractive models for studying the ability of the polymethylcarbosiloxane chain to form monomolecular layers on the surface of water. In the chains of these polymers, the SiOSi groups are only in the bridges connecting disilacycloalkanes. The π-
A isotherms of Polymers
X and
XI are shown in
Figure 10b and c. Polymer
X forms a stable monolayer which collapses at a surface area of 30 Å
2 per monomer unit and a surface pressure of 1.8 mN m
−1, whereas Polymer
XI does not spread over the surface of water at all. These differences in the surface properties between Polymers
X and
XI show that the ability of CL PMCS to form monomolecular films weakens with a decrease in the ratio of the number of hydrophilic to that of hydrophobic groups in the macromolecule. Comparing the behavior of Polymers
X and
XI, one should take into account not only the change in the O/CH
2 ratio, but also the change in the distance between the SiOSi groups in the backbone. By lengthening the hydrophobic segment from 2.63 Å in the 1,3-disilacyclobutane fragment of CL PMCS
X [
24] to 3.44 Å in the 1,4-disilacyclohexane moiety of CL PMCS
XI [
25] we violate the critical ratio that ensures the possibility for the monomolecular film to stay on the surface of water.
Figure 11 presents the dependence of the collapse surface pressure of a Langmuir monolayer on the ratio of the number of hydrophilic (oxygen atoms) to the number of hydrophobic (methyl and methylene groups) moieties in the monomer unit of CL PMCS
IV–
VI and
IX–
XI. For comparison, we also present the values of the collapse surface pressure of the monolayer π for CL PMS
I-
Me,
II-
Me, and two other CL PMS with asymmetric central fragment of the chain unit, namely, poly[oxy(2,4,4,6,8,8,10,10-octamethylcyclopentasiloxane-2,6-diyl)] (
XII-Me) and poly[oxy(2,4,4,6,6,8,10,10,12,12,14,14-dodecamethylcycloheptasiloxane-2,8-diyl)] (
XIII-Me) [
7]. From the data for the series of CL PMS
I-Me,
II-Me,
XII-Me, and
XIII-Me it follows that variation of the number of siloxane bonds in the polymer backbone (from cyclotetra- to cycloheptasiloxane) does not cause significant variations of the collapse surface pressure of monolayer. At the same time, partial replacement of siloxane bonds in the monomer unit by carbosilane bonds leads to a decrease in the collapse surface pressure of monolayer in proportion to the ratio of the number of hydrophilic to the number of hydrophobic groups in the monomer unit of CL PMCS.

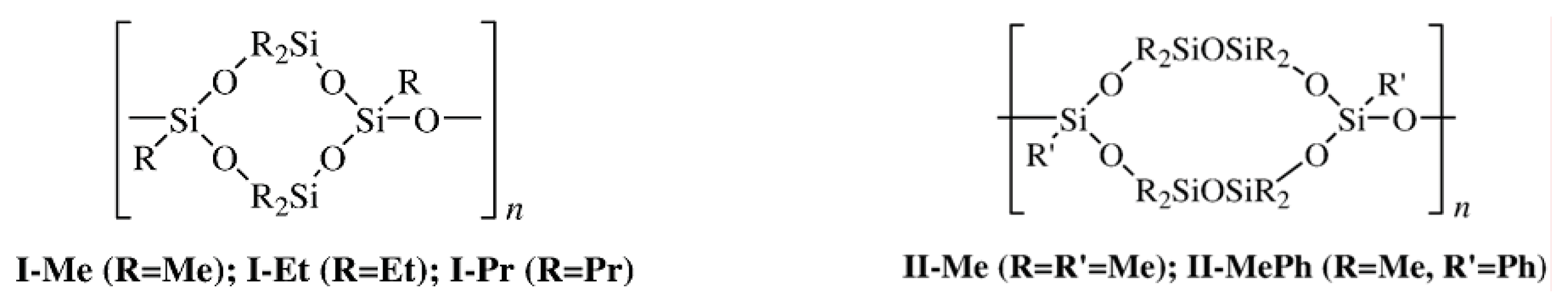




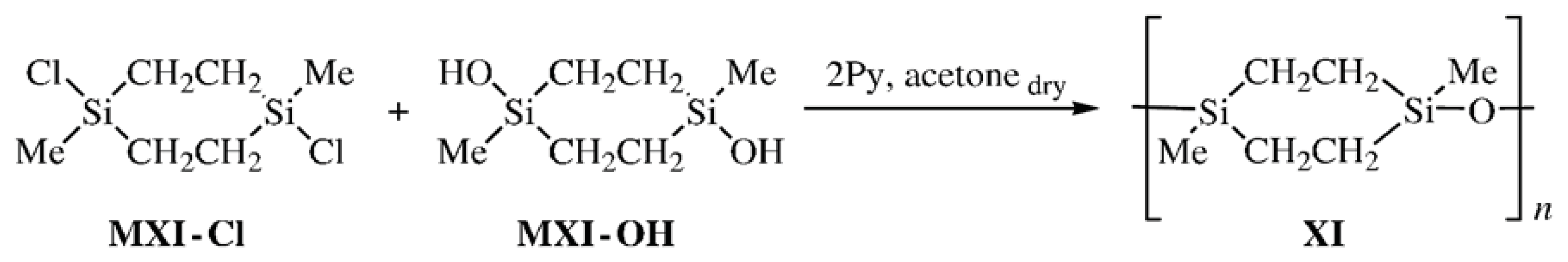

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}