Neuroprotective Effects of α-Tocotrienol on Kainic Acid-Induced Neurotoxicity in Organotypic Hippocampal Slice Cultures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
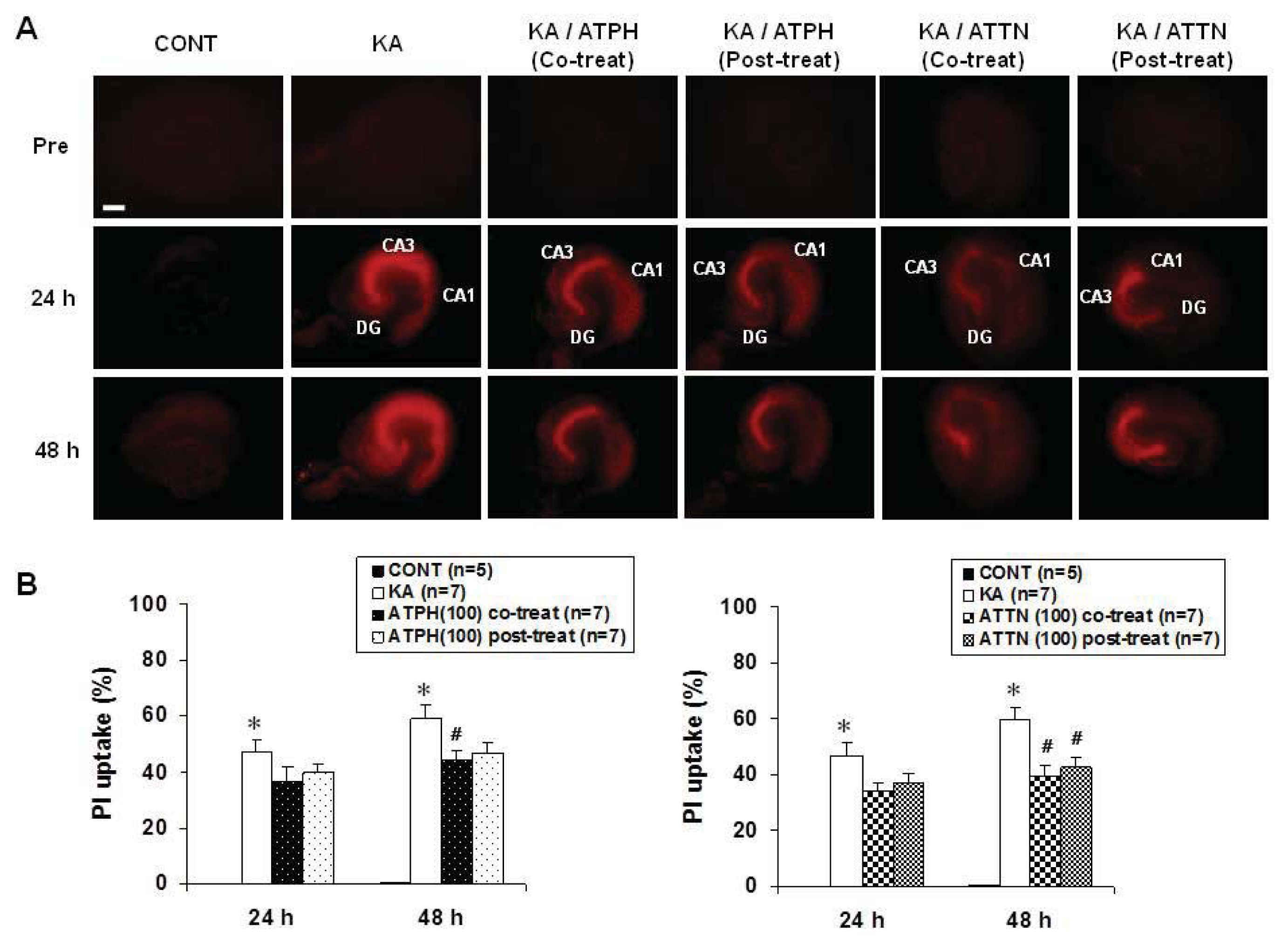
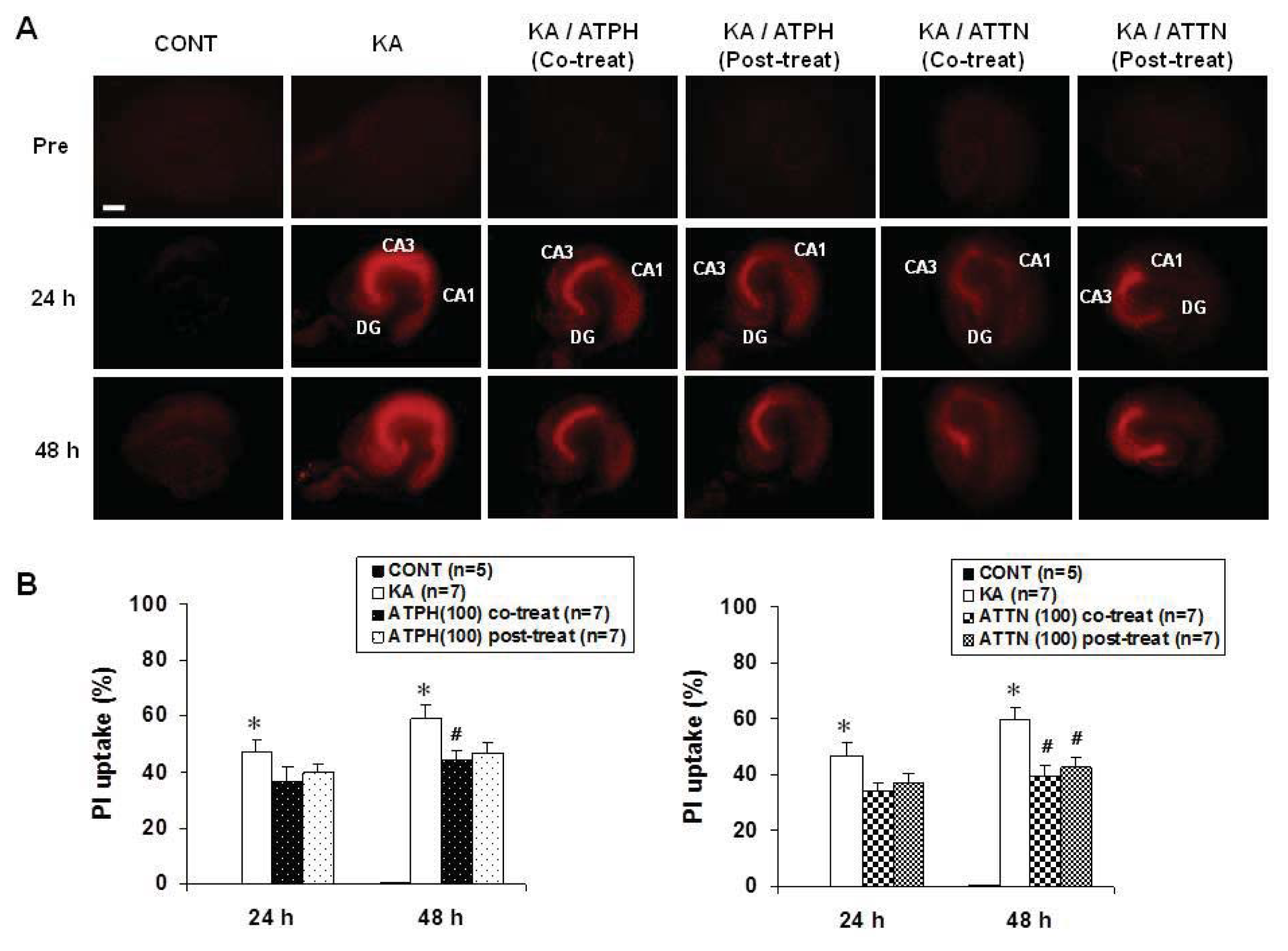
2.1. Effects of ATPH and ATTN on KA-Induced Neuronal Toxicity in OHSC
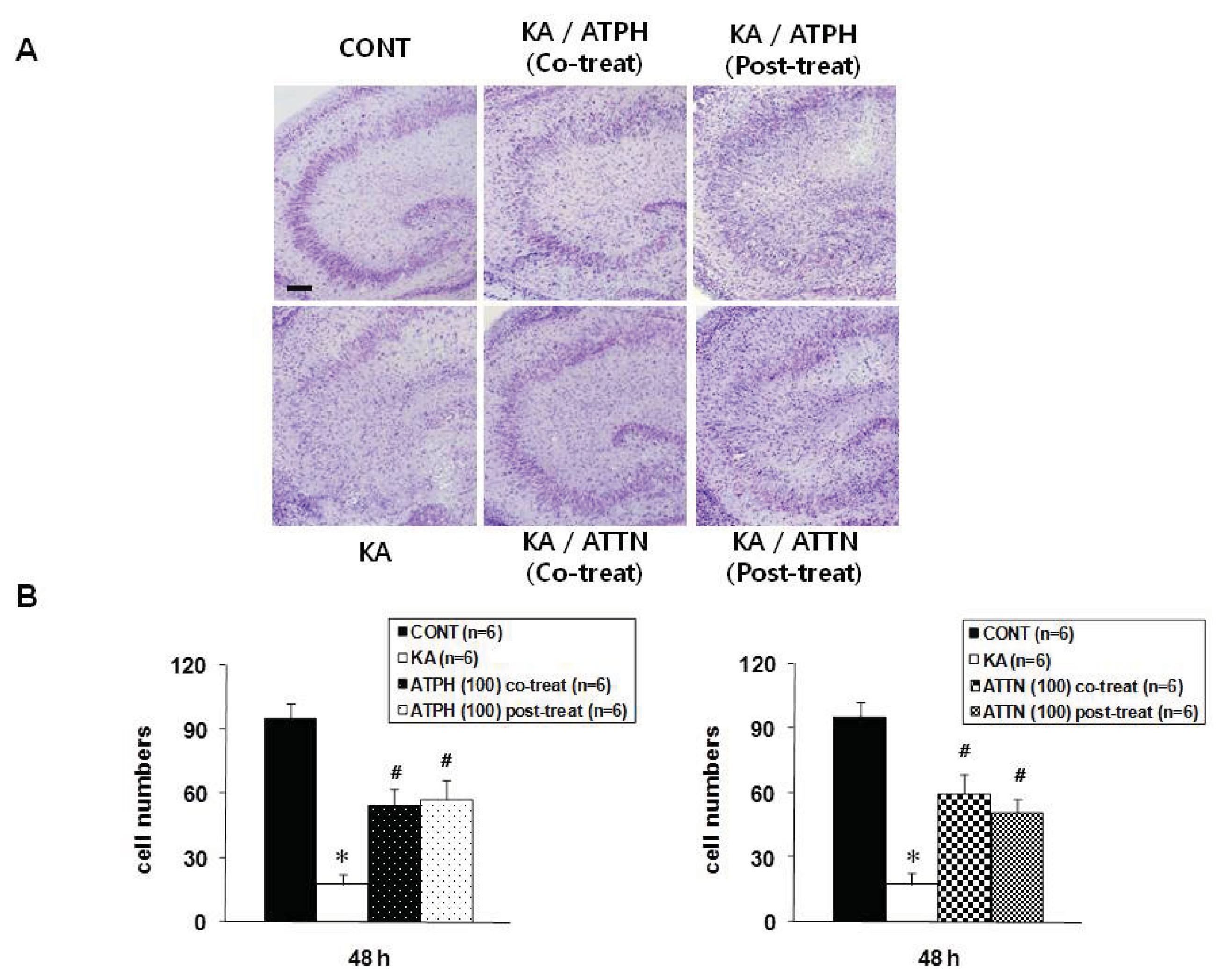
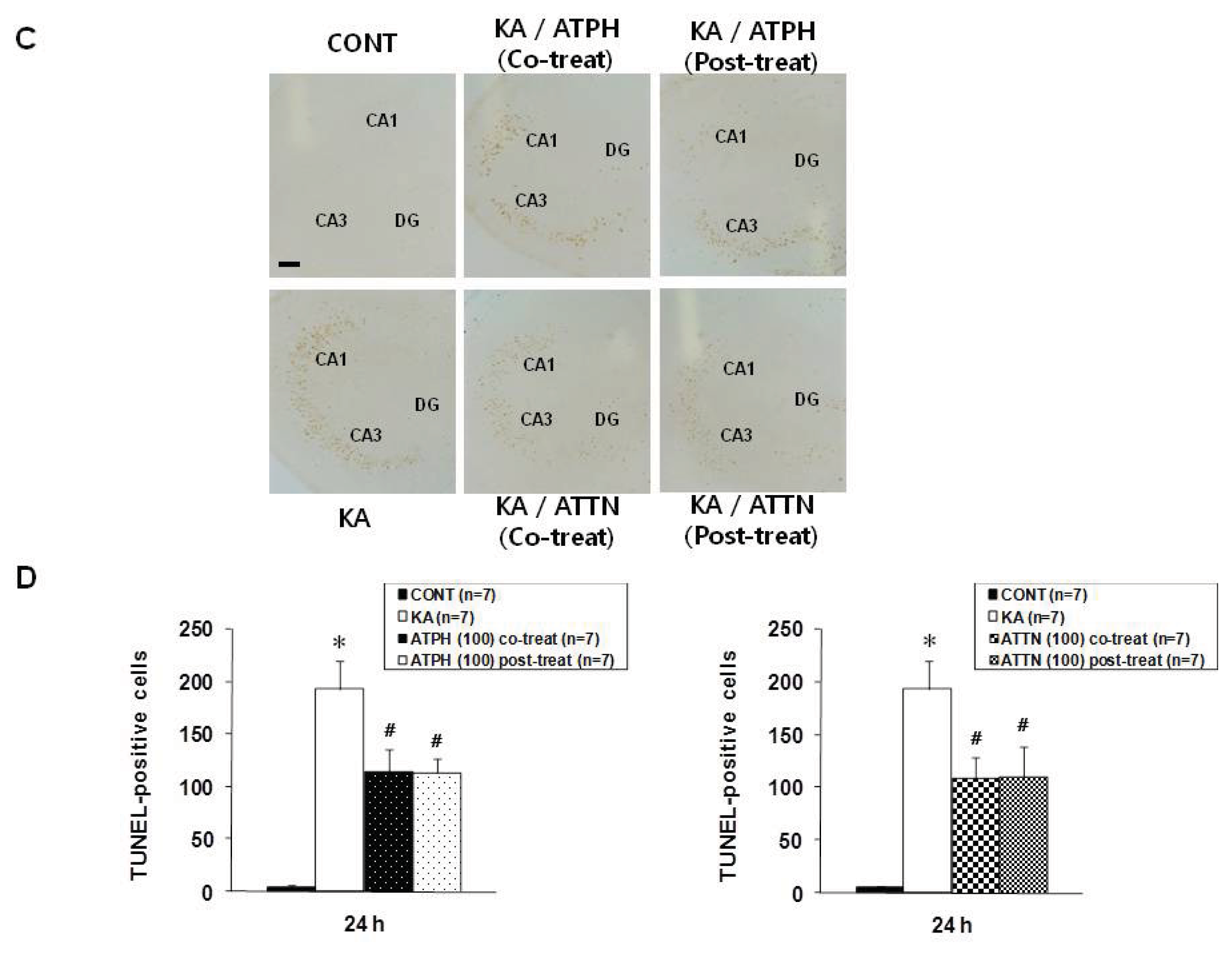
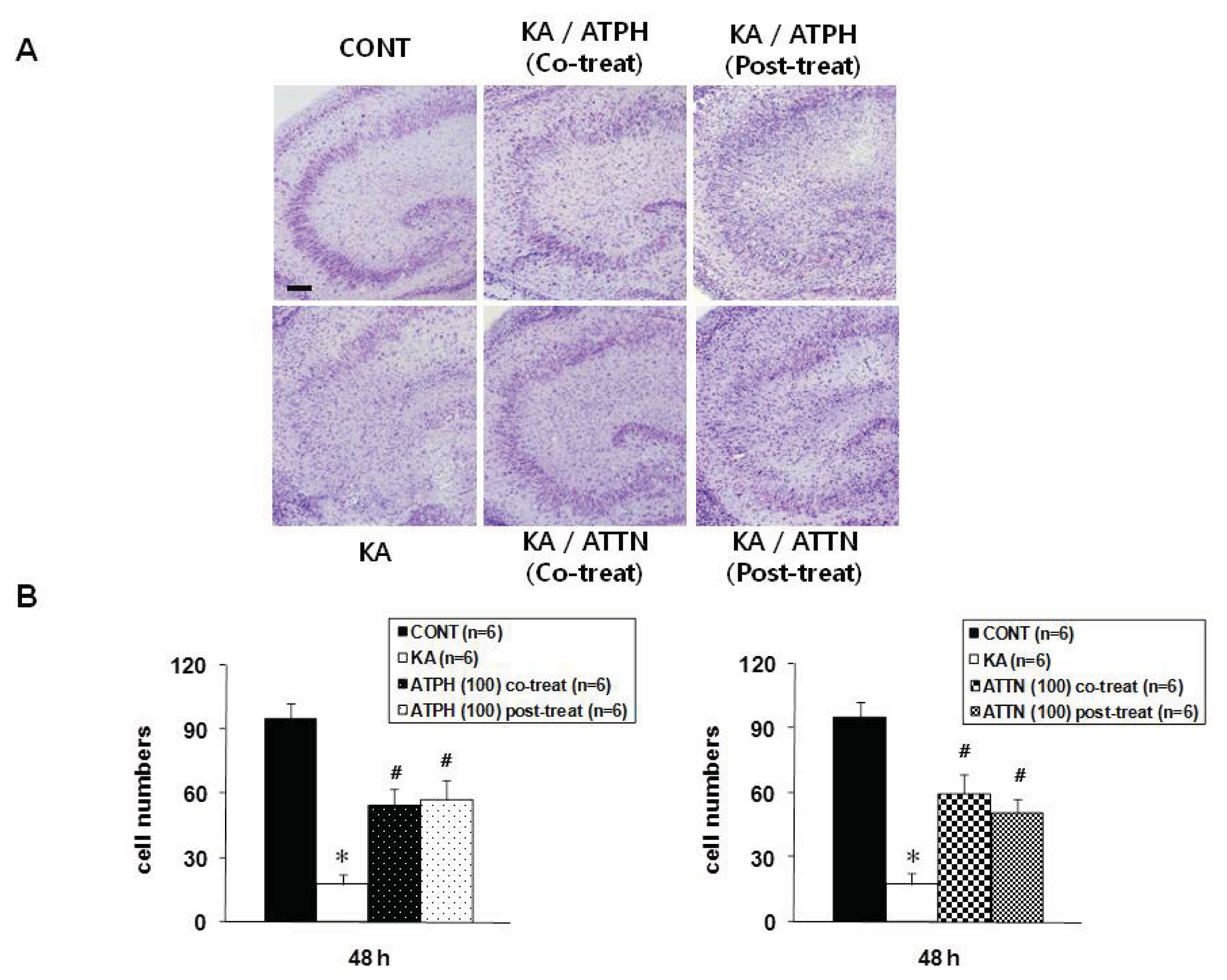
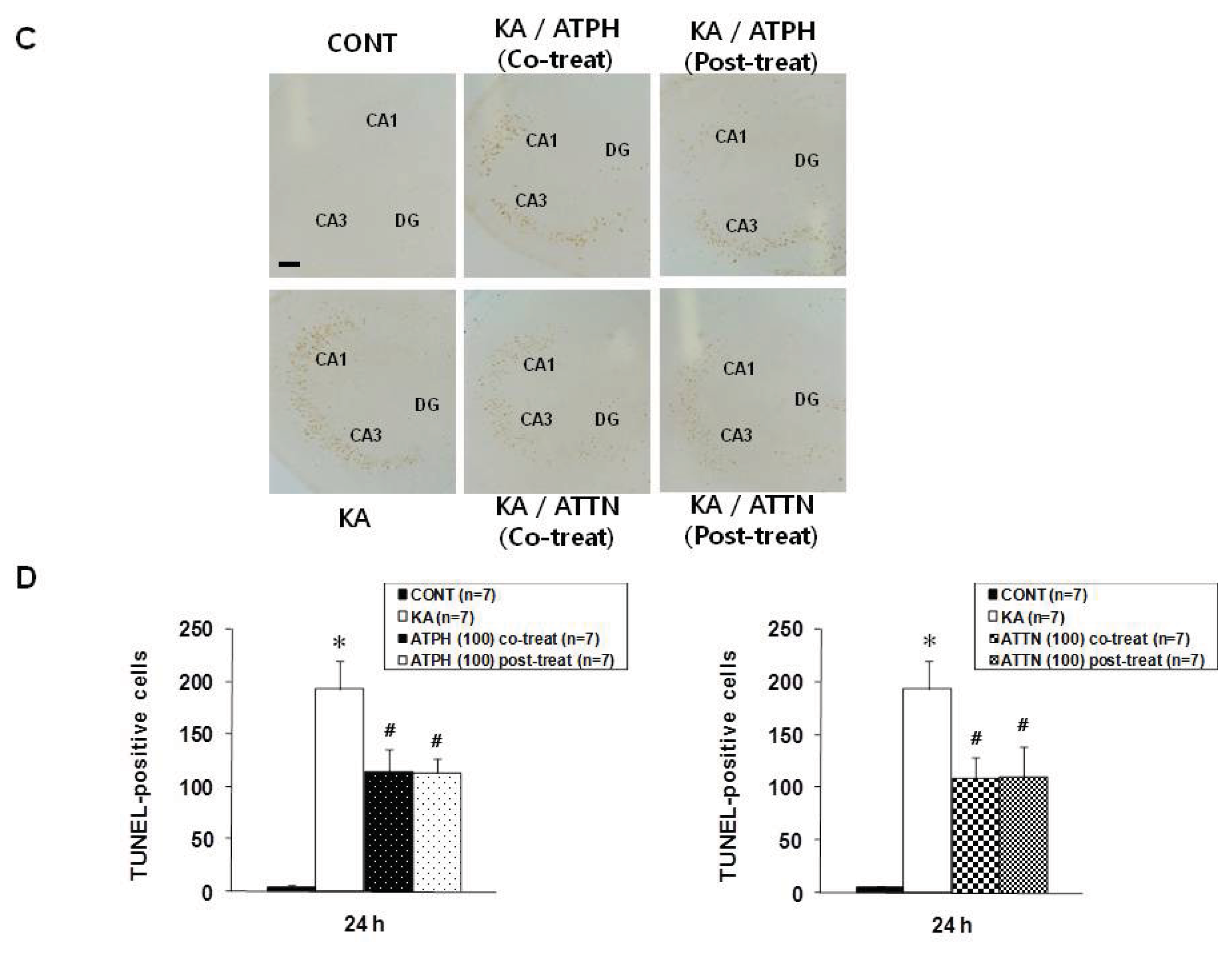
2.2. Cresyl Violet Staining and TUNEL Assay
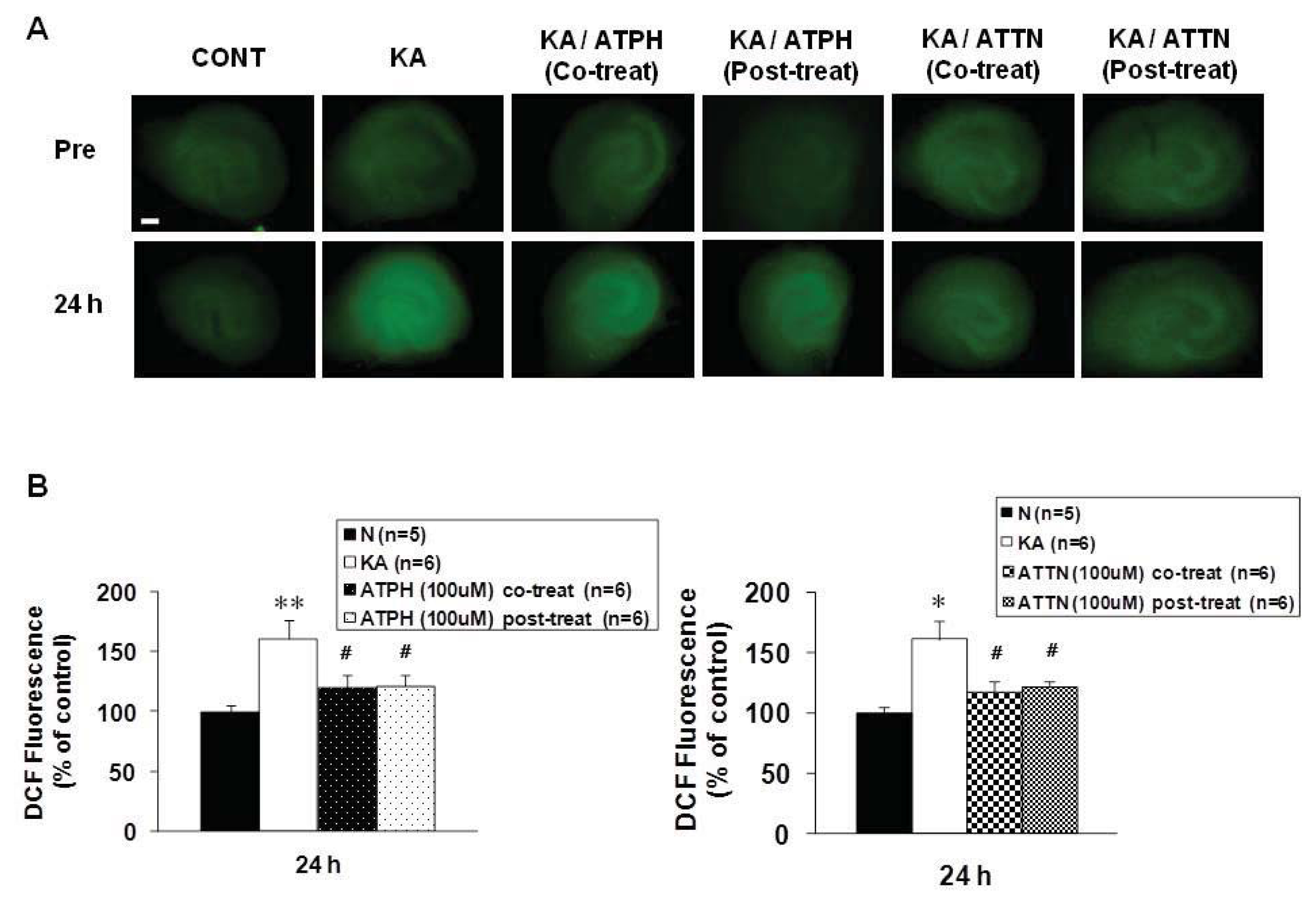
2.3. Formation of ROS in KA-Induced Toxicity
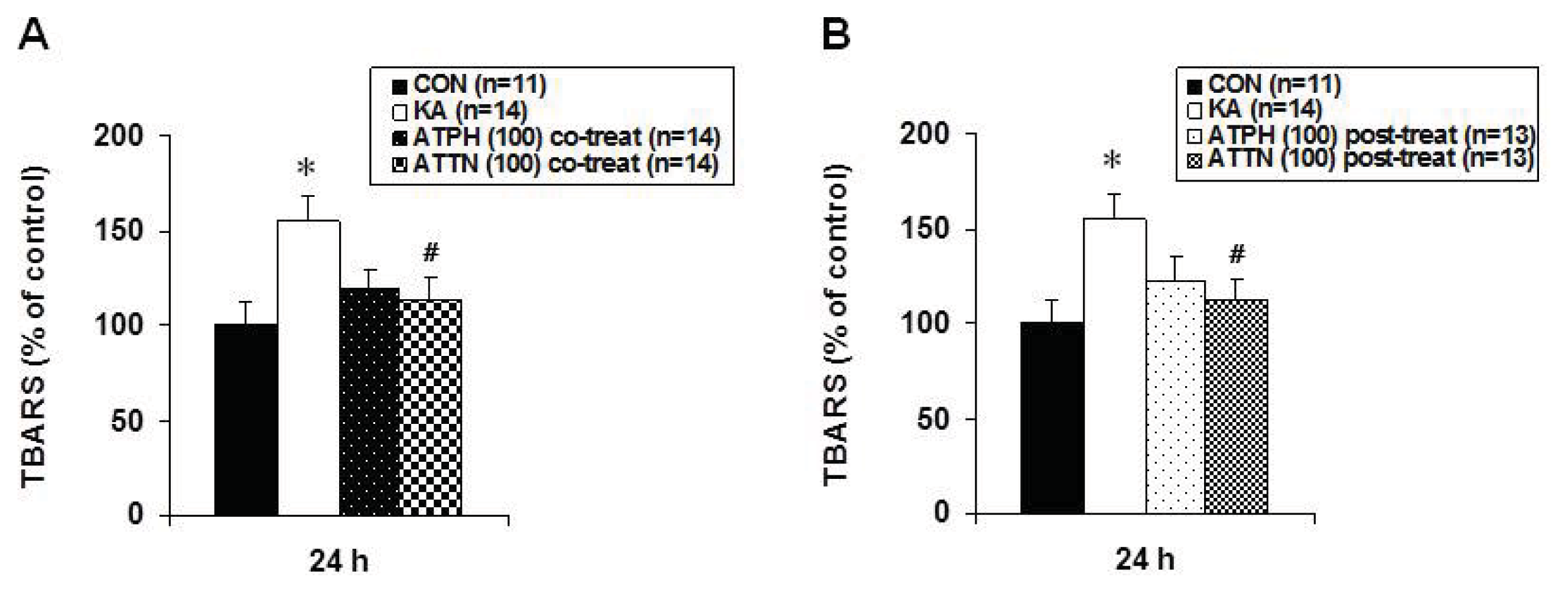
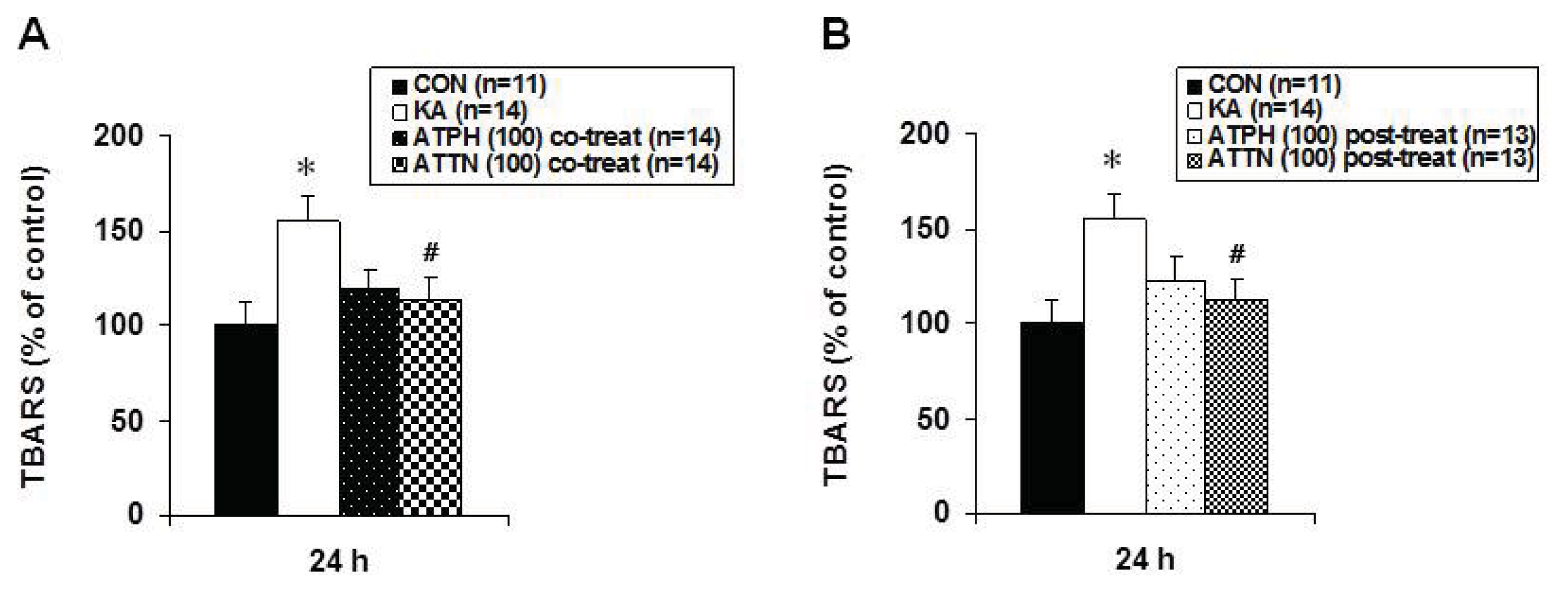
2.4. Attenuation of KA-Induced Lipid Peroxidation
2.5. Discussion
3. Experimental Section
3.1. Organotypic Slice Culture
3.2. Drug Treatment and Assessment of Neuronal Injury
3.3. Cresyl Violet Staining and TUNEL Assay
3.4. Evaluation of Intracellular ROS Formation
3.5. Lipid Peroxidation-TBARS Assay
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mohanan, P.V.; Yamamoto, H.A. Preventive effect of melatonin against brain mitochondria DNA damage, lipid peroxidation and seizures induced by kainic acid. Toxicol. Lett 2002, 129, 99–105. [Google Scholar]
- Lavrentiadou, S.N.; Tsantarliotou, M.P.; Zervos, I.A.; Nikolaidis, E.; Taitzoglou, I.A. CCl4 induces tissue-type plasminogen activator in rat brain; protective effects of oregano, rosemary or vitamin E. Food Chem. Toxicol. 2013, 4, S0278–S6915, , doi:10.1016/j.fct.2013.06.049.. [Google Scholar]
- Floyd, R.A. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc. Soc. Exp. Biol. Med 1999, 222, 236–245. [Google Scholar]
- Lee, H.; Jang, Y.H.; Lee, S.R. Protective effect of propofol against kainic acid-induced lipid peroxidation in mouse brain homogenates: Comparison with trolox and melatonin. J. Neurosurg. Anesthesiol 2005, 17, 144–148. [Google Scholar]
- Won, R.; Lee, K.H.; Lee, B.H. Coenzyme Q10 protects neurons against neurotoxicity in hippocampal slice culture. Neuroreport 2011, 22, 721–726. [Google Scholar]
- Kagan, V.E. Tocopherol stabilizes membrane against phospholipase A, free fatty acids, and lysophospholipids. Ann. N. Y. Acad. Sci 1989, 570, 121–135. [Google Scholar]
- Serbinova, E.A.; Packer, L. Antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Meth. Enzymol 1994, 234, 354–366. [Google Scholar]
- Hosomi, A.; Arita, M.; Sato, Y.; Kiyose, C.; Ueda, T.; Igarashi, O.; Arai, H.; Inoue, K. Affinity for alpha-tocopherol transfer protein as a determinant of the biological activities of vitamin E analogs. FEBS Lett 1997, 409, 105–108. [Google Scholar]
- Uchida, T.; Nomura, S.; Ichikawa, T.; Abe, C.; Ikeda, S. Tissue distribution of vitamin E metabolites in rats after oral administration of tocopherol or tocotrienol. J. Nutr. Sci. Vitaminol. 2011, 57, 326–332. [Google Scholar]
- Tomeo, A.C.; Geller, M.; Watkins, T.R.; Gapor, A.; Bierenbaum, M.L. Antioxidant effects of tocotrienols in patients with hyperlipidemia and carotid stenosis. Lipids 1995, 30, 1179–1183. [Google Scholar]
- Parker, R.A.; Pearce, B.C.; Clark, R.W.; Gordon, D.A.; Wright, J.J. Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem 1993, 268, 11230–11238. [Google Scholar]
- Guthrie, N.; Gapor, A.; Chambers, A.F.; Carroll, K.K. Inhibition of proliferation of estrogen receptor-negative MDA-MB-435 and -positive MCF-7 human breast cancer cells by palm oil tocotrienols and tamoxifen, alone and in combination. J. Nutr 1997, 127, 544S–548S. [Google Scholar]
- Nesaretnam, K.; Stephen, R.; Dils, R.; Darbre, P. Tocotrienols inhibit the growth of human breast cancer cells irrespective of estrogen receptor status. Lipids 1998, 33, 461–469. [Google Scholar]
- Numakawa, Y.; Numakawa, T.; Matsumoto, T.; Yagasak, I.Y.; Kumamaru, E.; Kunugi, H.; Taguchi, T.; Niki, E. Vitamin E protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. J. Neurochem. 2006, 97, 1191–1202. [Google Scholar]
- Miyamoto, K.; Shiozaki, M.; Shibata, M.; Koike, M.; Uchiyama, Y.; Gotow, T. Very-high-dose alpha-tocopherol supplementation increases blood pressure and causes possible adverse central nervous system effects in stroke-prone spontaneously hypertensive rats. J. Neurosci. Res 2009, 87, 556–566. [Google Scholar]
- Gahwiler, B.H.; Capogna, M.; Debanne, D.; McKinney, R.A.; Thompson, S.M. Organotypic slice cultures: A technique has come of age. Trends Neurosci 1997, 20, 471–477. [Google Scholar]
- Lindroos, M.M.; Soini, S.L.; Kukko-Lukjanov, T.K.; Korpi, E.R.; Lovinger, D.; Holopainen, I.E. Maturation of cultured hippocampal slices results in increased excitability in granule cells. Int. J. Dev. Neurosci 2005, 23, 65–73. [Google Scholar]
- Lee, K.H.; Park, J.H.; Won, R.; Lee, H.J.; Nam, T.S.; Lee, B.H. Inhibition of hexokinase leads to neuroprotection against excitotoxicity in organotypic hippocampal slice culture. J. Neurosci. Res 2011, 89, 96–107. [Google Scholar]
- Nadler, J.V. Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci 1981, 29, 2031–2042. [Google Scholar]
- Okazaki, M.M.; Nadler, J.V. Protective effects of mossy fiber lesions against kainic acid-induced seizures and neuronal degeneration. Neuroscience 1988, 26, 763–781. [Google Scholar]
- Caeser, M.; Aertsen, A. Morphological organization of rat hippocampal slice cultures. J. Comp. Neurol. 1991, 307, 87–106. [Google Scholar]
- Lee, K.H.; Won, R.; Kim, U.J.; Kim, G.M.; Chung, M.A.; Sohn, J.H.; Lee, B.H. Neuroprotective effects of FK506 against excitotoxicity in organotypic hippocampal slice culture. Neurosci. Lett 2010, 474, 126–130. [Google Scholar]
- Leist, M.; Jaattela, M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol 2001, 2, 589–598. [Google Scholar]
- Kim, E.J.; Won, R.; Sohn, J.H.; Chung, M.A.; Nam, T.S.; Lee, H.J.; Lee, B.H. Anti-oxidant effect of ascorbic and dehydroascorbic acids in hippocampal slice culture. Biochem. Biophys. Res. Commun 2008, 366, 8–14. [Google Scholar]
- Bromont, C.; Marie, C.; Bralet, J. Increased lipid peroxidation in vulnerable brain regions after transient forebrain ischemia in rats. Stroke 1989, 20, 918–924. [Google Scholar]
- Sharma, S.S.; Kaundal, R.K. Neuroprotective effects of 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox), an antioxidant in middle cerebral artery occlusion induced focal cerebral ischemia in rats. Neurol. Res 2007, 29, 304–309. [Google Scholar]
- Liu, R.; Liu, W.; Doctrow, S.R.; Baudry, M. Iron toxicity in organotypic cultures of hippocampal slices: Role of reactive oxygen species. J. Neurochem 2003, 85, 492–502. [Google Scholar]
- Levy, S.L.; Burnham, W.M.; Hwang, P.A. An evaluation of the anticonvulsant effects of vitamin E. Epilepsy Res 1990, 6, 12–17. [Google Scholar]
- Levy, S.L.; Burnham, W.M.; Bishai, A.; Hwang, P.A. The anticonvulsant effects of vitamin E: A further evaluation. Can. J. Neurol. Sci 1992, 19, 201–203. [Google Scholar]
- Burton, G.W.; Joyce, A.; Ingold, K.U. Is vitamin E the only lipid-soluble, chain-breaking antioxidant in human blood plasma and erythrocyte membranes? Arch. Biochem. Biophys 1983, 221, 281–290. [Google Scholar]
- Cooney, R.V.; Franke, A.A.; Harwood, P.J.; Hatch-Pigott, V.; Custer, L.J.; Mordan, L.J. Gamma-tocopherol detoxification of nitrogen dioxide: Superiority to alpha-tocopherol. Proc. Natl. Acad. Sci. USA 1993, 90, 1771–1775. [Google Scholar]
- Serbinova, E.; Kagan, V.; Han, D.; Packer, L. Free radical recycling and intramembrane mobility in the antioxidant properties of alpha-tocopherol and alpha-tocotrienol. Free Radic. Biol. Med 1991, 10, 263–275. [Google Scholar]
- Suzuki, Y.J.; Tsuchiya, M.; Wassall, S.R.; Choo, Y.M.; Govil, G.; Kagan, V.E.; Packer, L. Structural and dynamic membrane properties of alpha-tocopherol and alpha-tocotrienol: Implication to the molecular mechanism of their antioxidant potency. Biochemistry 1993, 32, 10692–10699. [Google Scholar]
- Kamat, J.P.; Devasagayam, T.P. Tocotrienols from palm oil as potent inhibitors of lipid peroxidation and protein oxidation in rat brain mitochondria. Neurosci. Lett 1995, 195, 179–182. [Google Scholar]
- Schneider, C. Chemistry and biology of vitamin E. Mol. Nutr. Food Res 2005, 49, 7–30. [Google Scholar]
- Niki, E.; Saito, T.; Kawakami, A.; Kamiya, Y. Inhibition of oxidation of methyl linoleate in solution by vitamin E and vitamin C. J. Biol. Chem 1984, 259, 4177–4182. [Google Scholar]
- Shi, H.; Noguchi, N.; Niki, E. Comparative study on dynamics of antioxidative action of alpha-tocopheryl hydroquinone, ubiquinol, and alpha-tocopherol against lipid peroxidation. Free Radic. Biol. Med 1999, 27, 334–346. [Google Scholar]
- Stoppini, L.; Buchs, P.A.; Muller, D.A. Simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 1991, 37, 173–182. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jung, N.Y.; Lee, K.H.; Won, R.; Lee, B.H. Neuroprotective Effects of α-Tocotrienol on Kainic Acid-Induced Neurotoxicity in Organotypic Hippocampal Slice Cultures. Int. J. Mol. Sci. 2013, 14, 18256-18268. https://doi.org/10.3390/ijms140918256
Jung NY, Lee KH, Won R, Lee BH. Neuroprotective Effects of α-Tocotrienol on Kainic Acid-Induced Neurotoxicity in Organotypic Hippocampal Slice Cultures. International Journal of Molecular Sciences. 2013; 14(9):18256-18268. https://doi.org/10.3390/ijms140918256
Chicago/Turabian StyleJung, Na Young, Kyung Hee Lee, Ran Won, and Bae Hwan Lee. 2013. "Neuroprotective Effects of α-Tocotrienol on Kainic Acid-Induced Neurotoxicity in Organotypic Hippocampal Slice Cultures" International Journal of Molecular Sciences 14, no. 9: 18256-18268. https://doi.org/10.3390/ijms140918256