Oxidative Stress in the Pathogenesis of Keratoconus and Fuchs Endothelial Corneal Dystrophy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
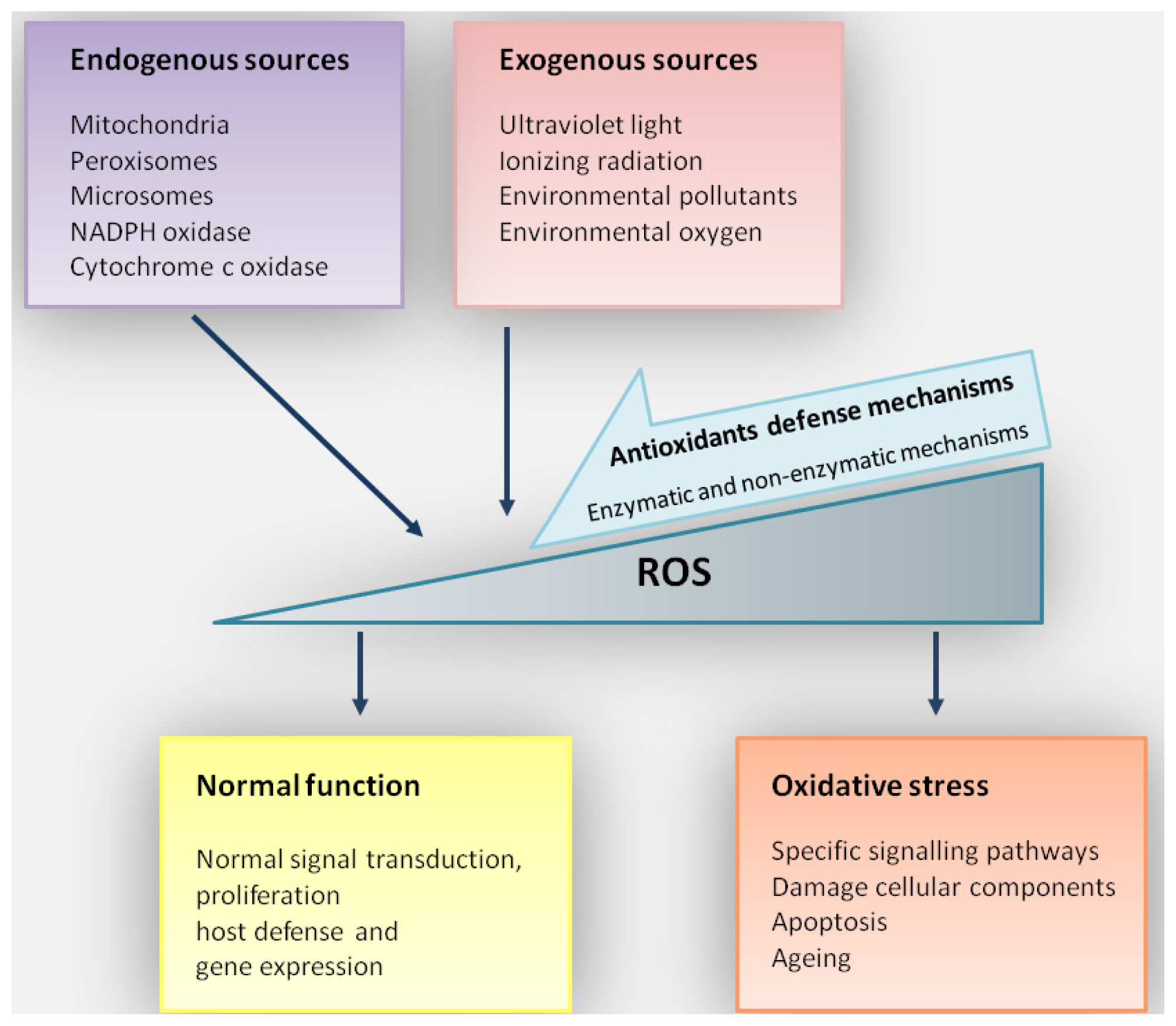
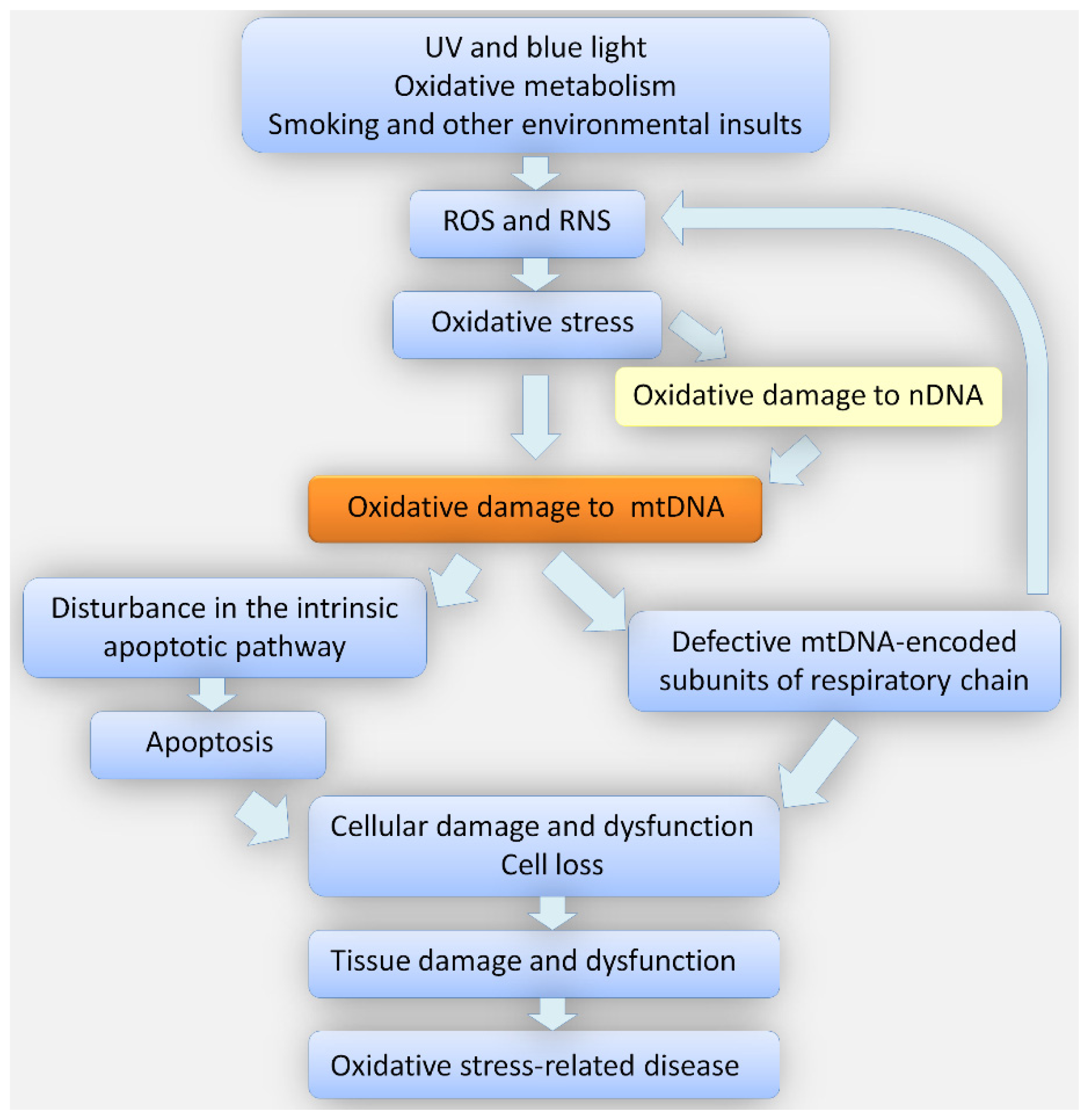
2. Oxidative Stress
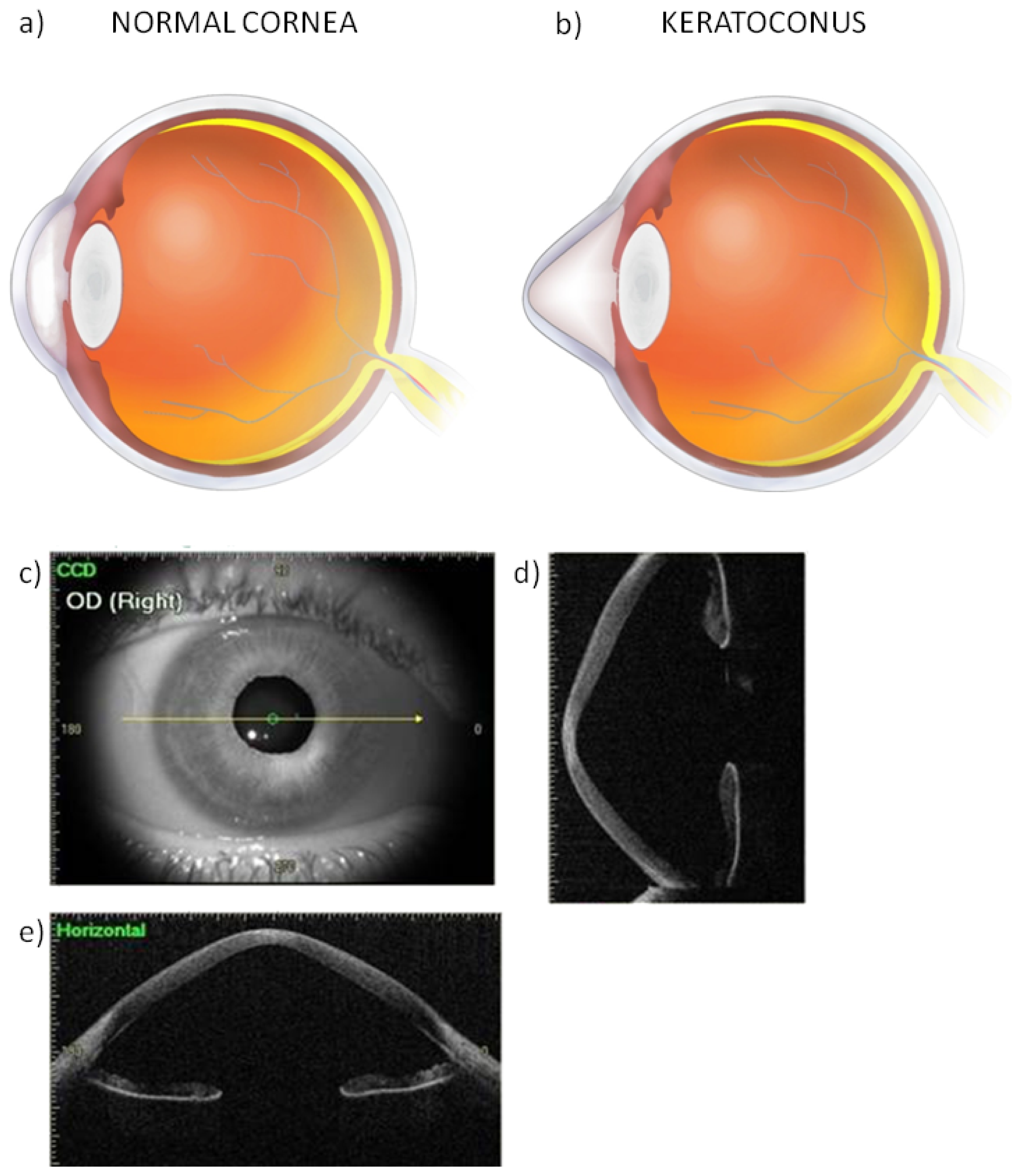
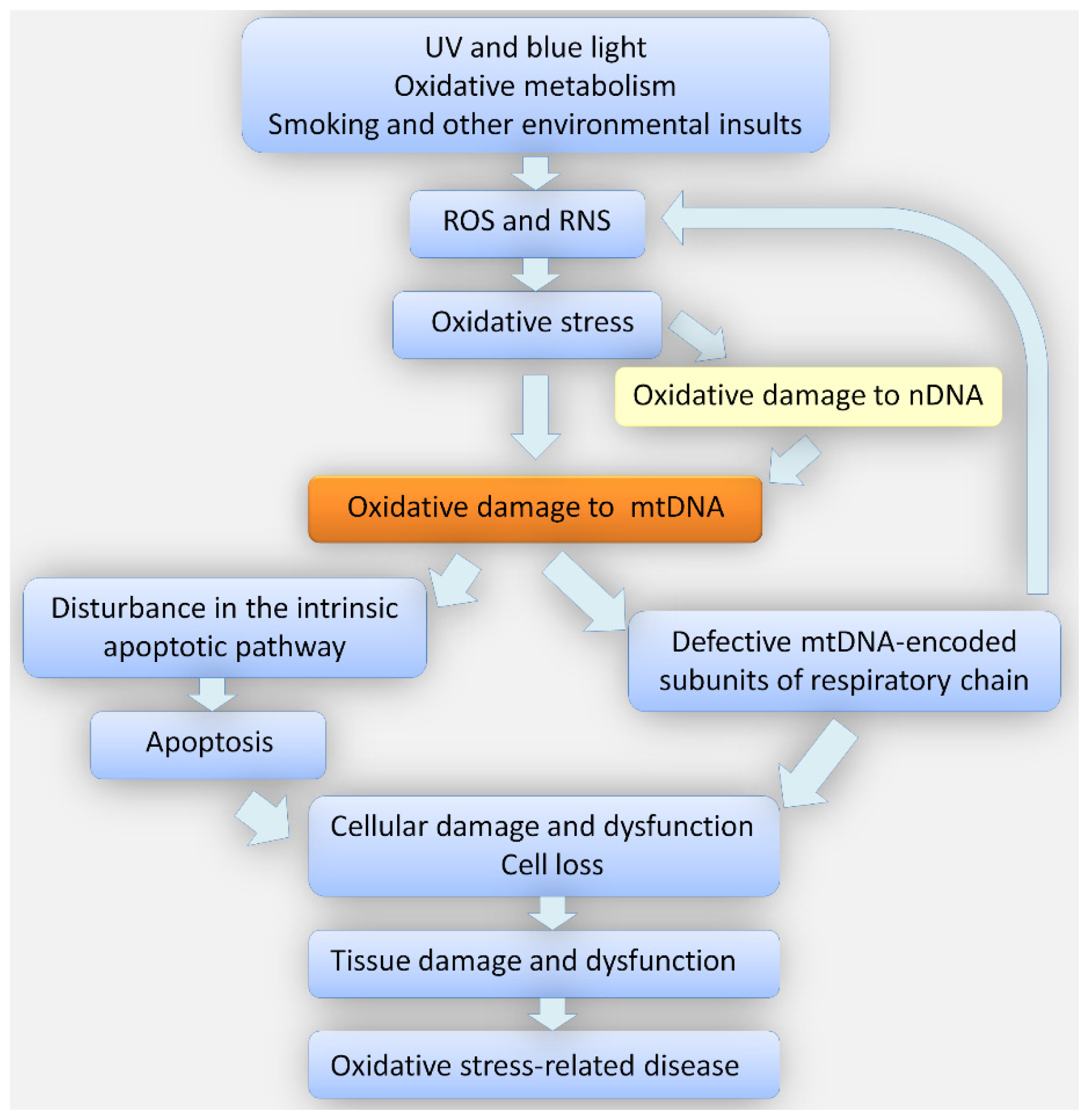
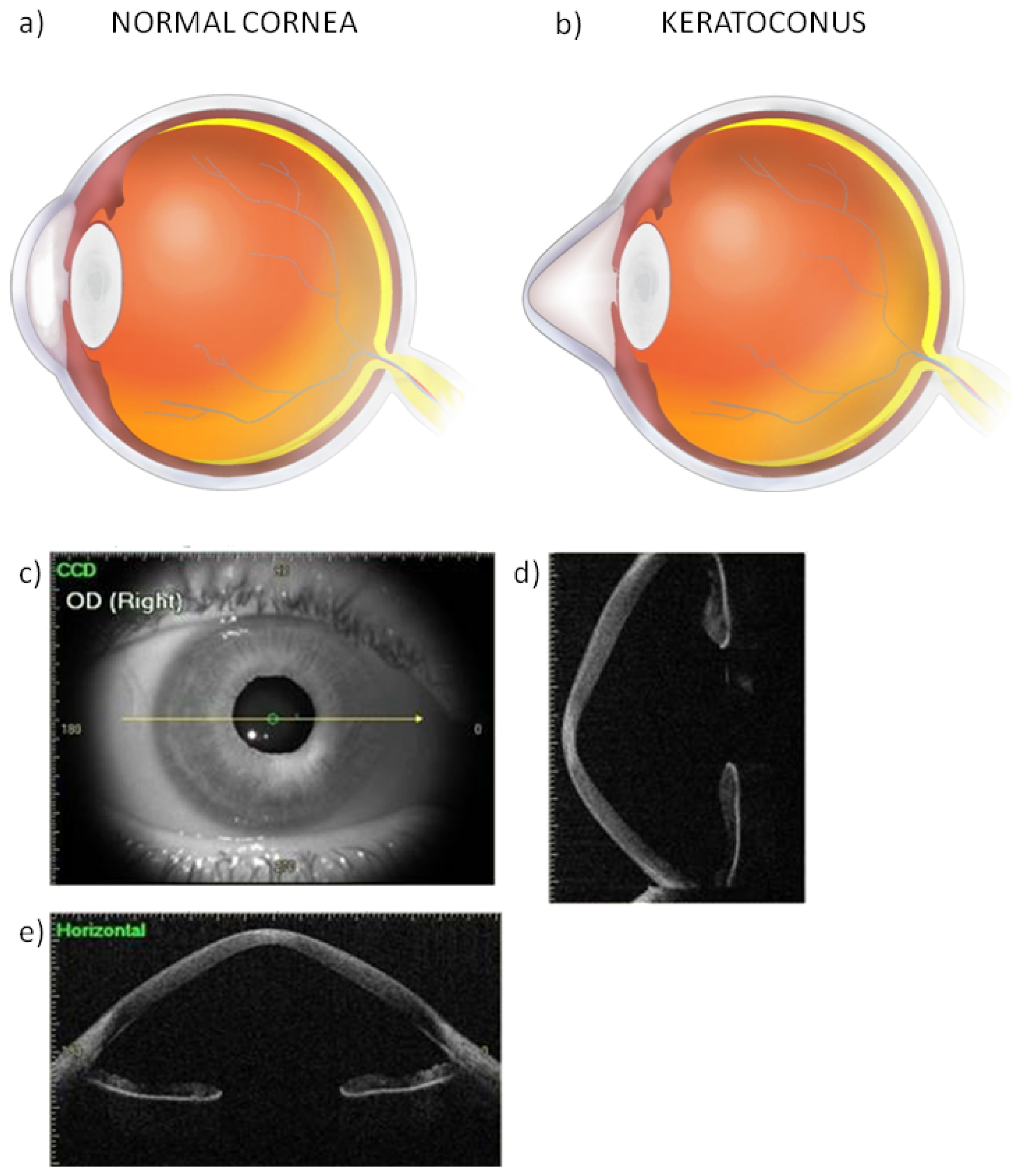
3. Oxidative Stress in the Pathogenesis of Keratoconus
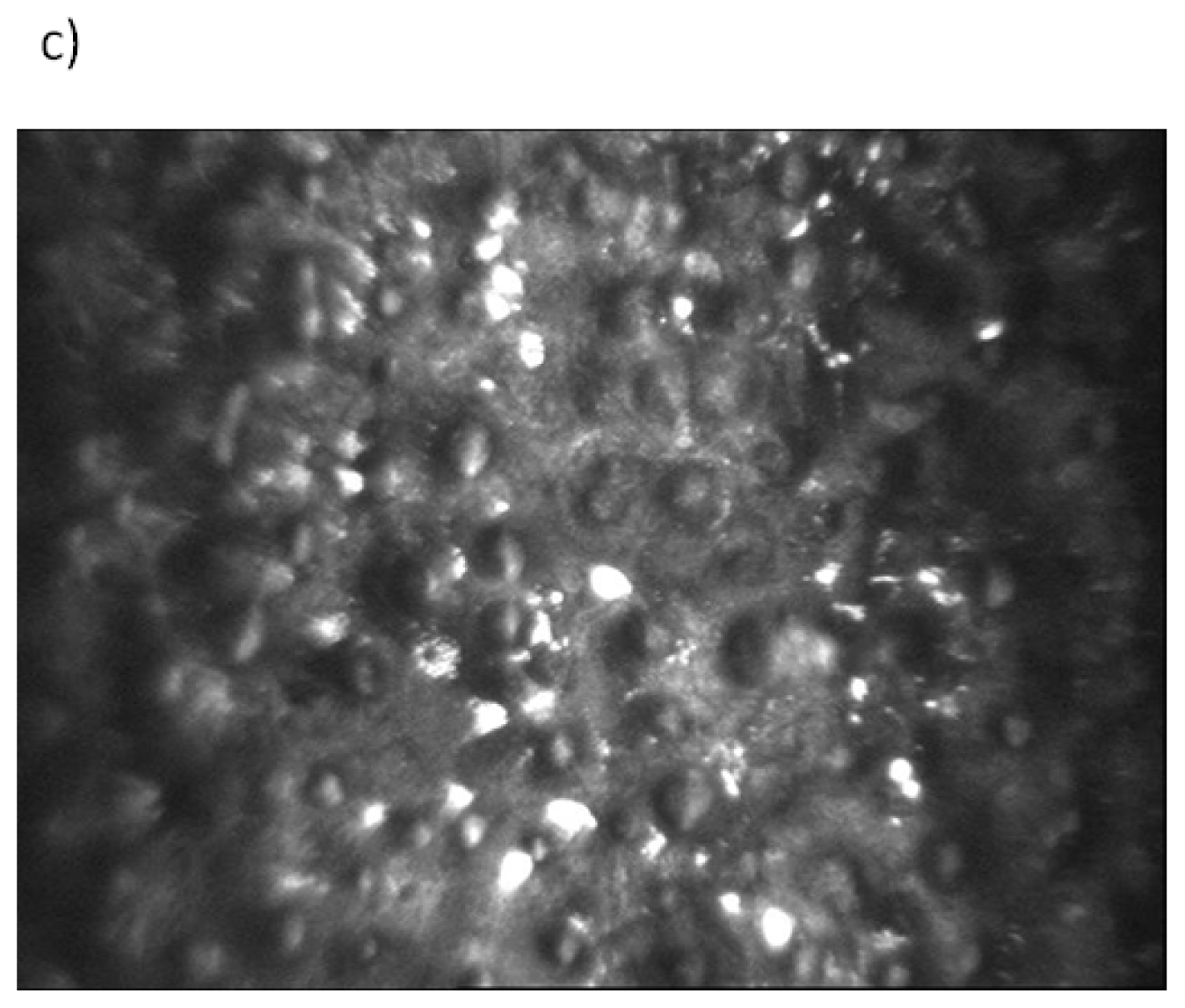
4. Oxidative Stress in the Pathogenesis of Fuchs Endothelial Corneal Dystrophy
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Choi, S.I.; Dadakhujaev, S.; Ryu, H.; Im Kim, T.; Kim, E.K. Melatonin protects against oxidative stress in granular corneal dystrophy type 2 corneal fibroblasts by mechanisms that involve membrane melatonin receptors. J. Pineal Res 2011, 51, 94–103. [Google Scholar]
- Greinert, R.; Volkmer, B.; Henning, S.; Breitbart, E.W.; Greulich, K.O.; Cardoso, M.C.; Rapp, A. UVA-induced DNA double-strand breaks result from the repair of clustered oxidative DNA damages. Nucleic Acids Res 2012, 40, 10263–10273. [Google Scholar]
- Cai, C.X.; Birk, D.E.; Linsenmayer, T.F. Nuclear ferritin protects DNA from UV damage in corneal epithelial cells. Mol. Biol. Cell 1998, 9, 1037–1051. [Google Scholar]
- Chen, Y.; Mehta, G.; Vasiliou, V. Antioxidant defenses in the ocular surface. Ocul. Surf 2009, 7, 176–185. [Google Scholar]
- Chai, D.; Gaster, R.N.; Roizenblatt, R.; Juhasz, T.; Brown, D.J.; Jester, J.V. Quantitative assessment of UVA-riboflavin corneal cross-linking using nonlinear optical microscopy. Invest. Ophthalmol. Vis. Sci 2011, 52, 4231–4238. [Google Scholar]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J 2012, 5, 9–19. [Google Scholar]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res 2011, 711, 193–201. [Google Scholar]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact 2006, 160, 1–40. [Google Scholar]
- Kenney, M.C.; Chwa, M.; Atilano, S.R.; Tran, A.; Carballo, M.; Saghizadeh, M.; Vasiliou, V.; Adachi, W.; Brown, D.J. Increased levels of catalase and cathepsin V/L2 but decreased TIMP-1 in keratoconus corneas: Evidence that oxidative stress plays a role in this disorder. Invest. Ophthalmol. Vis. Sci 2005, 46, 823–832. [Google Scholar]
- Chwa, M.; Atilano, S.R.; Hertzog, D.; Zheng, H.; Langberg, J.; Kim, D.W.; Kenney, M.C. Hypersensitive response to oxidative stress in keratoconus corneal fibroblasts. Invest. Ophthalmol. Vis. Sci 2008, 49, 4361–4369. [Google Scholar]
- Jurkunas, U.V.; Bitar, M.S.; Funaki, T.; Azizi, B. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am. J. Pathol 2010, 177, 2278–2289. [Google Scholar]
- Nakamura, S.; Shibuya, M.; Nakashima, H.; Hisamura, R.; Masuda, N.; Imagawa, T.; Uehara, M.; Tsubota, K. Involvement of oxidative stress on corneal epithelial alterations in a blink-suppressed dry eye. Invest. Ophthalmol. Vis. Sci 2007, 48, 1552–1558. [Google Scholar]
- Marak, G.E.; de Kozak, Y.; Faure, J.P. Free radicals and antioxidants in the pathogenesis of eye diseases. Adv. Exp. Med. Biol 1990, 264, 513–527. [Google Scholar]
- Fang, Y.Z.; Yang, S.; Wu, G. Free radicals, antioxidants, and nutrition. Nutrition 2002, 18, 872–879. [Google Scholar]
- Ishii, T.; Miyazawa, M.; Onouchi, H.; Yasuda, K.; Hartman, P.S.; Ishii, N. Model animals for the study of oxidative stress from complex II. Biochim. Biophys. Acta 2013, 1827, 588–597. [Google Scholar]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox. Signal 2008, 10, 1343–1374. [Google Scholar]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol 2002, 30, 620–650. [Google Scholar]
- Sheu, S.S.; Nauduri, D.; Anders, M.W. Targeting antioxidants to mitochondria: A new therapeutic direction. Biochim. Biophys. Acta 2006, 1762, 256–265. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol 1997, 82, 291–295. [Google Scholar]
- De Bonis, P.; Laborante, A.; Pizzicoli, C.; Stallone, R.; Barbano, R.; Longo, C.; Mazzilli, E.; Zelante, L.; Bisceglia, L. Mutational screening of VSX1, SPARC, SOD1, LOX, and TIMP3 in keratoconus. Mol. Vis 2011, 17, 2482–2494. [Google Scholar]
- Rabinowitz, Y.S. Keratoconus. Surv. Ophthalmol 1998, 42, 297–319. [Google Scholar]
- Romero-Jiménez, M.; Santodomingo-Rubido, J.; Wolffsohn, J.S. Keratoconus: A review. Cont. Lens Anterior Eye 2010, 33, 157–166. [Google Scholar]
- Kim, W.J.; Rabinowitz, Y.S.; Meisler, D.M.; Wilson, S.E. Keratocyte apoptosis associated with keratoconus. Exp. Eye Res 1999, 69, 475–481. [Google Scholar]
- Ahmadi Hosseini, S.M.; Mohidin, N.; Abolbashari, F.; Mohd-Ali, B.; Santhirathelagan, C.T. Corneal thickness and volume in subclinical and clinical keratoconus. Int. Ophthalmol 2013, 33, 139–145. [Google Scholar]
- Sherwin, T.; Brookes, N.H. Morphological changes in keratoconus: Pathology or pathogenesis. Clin. Exp. Ophthalmol 2004, 32, 211–217. [Google Scholar]
- Burdon, K.P.; Vincent, A.L. Insights into keratoconus from a genetic perspective. Clin. Exp. Optom 2013, 96, 146–154. [Google Scholar]
- Edwards, M.; McGhee, C.N.; Dean, S. The genetics of keratoconus. Clin. Exp. Ophthalmol 2001, 29, 345–351. [Google Scholar]
- Kenney, M.C.; Brown, D.J. The cascade hypothesis of keratoconus. Cont. Lens Anterior Eye 2003, 26, 139–146. [Google Scholar]
- Saee-Rad, S.; Hashemi, H.; Miraftab, M.; Noori-Daloii, M.R.; Chaleshtori, M.H.; Raoofian, R.; Jafari, F.; Greene, W.; Fakhraie, G.; Rezvan, F.; et al. Mutation analysis of VSX1 and SOD1 in Iranian patients with keratoconus. Mol. Vis 2011, 17, 3128–3136. [Google Scholar]
- Tanwar, M.; Kumar, M.; Nayak, B.; Pathak, D.; Sharma, N.; Titiyal, J.S.; Dada, R. VSX1 gene analysis in keratoconus. Mol. Vis 2010, 16, 2395–2401. [Google Scholar]
- Stabuc-Silih, M.; Strazisar, M.; Ravnik-Glavac, M.; Hawlina, M.; Glavac, D. Genetics and clinical characteristics of keratoconus. Acta Dermatovenerol. Alp. Panonica Adriat 2010, 19, 3–10. [Google Scholar]
- Stabuc-Silih, M.; Ravnik-Glavac, M.; Glavac, D.; Hawlina, M.; Strazisar, M. Polymorphisms in COL4A3 and COL4A4 genes associated with keratoconus. Mol. Vis 2009, 15, 2848–2860. [Google Scholar]
- Hughes, A.E.; Bradley, D.T.; Campbell, M.; Lechner, J.; Dash, D.P.; Simpson, D.A.; Willoughby, C.E. Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am. J. Hum. Genet 2011, 89, 628–633. [Google Scholar]
- Chwa, M.; Atilano, S.R.; Reddy, V.; Jordan, N.; Kim, D.W.; Kenney, M.C. Increased stress-induced generation of reactive oxygen species and apoptosis in human keratoconus fibroblasts. Invest. Ophthalmol. Vis. Sci 2006, 47, 1902–1910. [Google Scholar]
- Buddi, R.; Lin, B.; Atilano, S.R.; Zorapapel, N.C.; Kenney, M.C.; Brown, D.J. Evidence of oxidative stress in human corneal diseases. J. Histochem. Cytochem 2002, 50, 341–351. [Google Scholar]
- Squadrito, G.L.; Pryor, W.A. Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic. Biol. Med 1998, 25, 392–403. [Google Scholar]
- Arnal, E.; Peris-Martínez, C.; Menezo, J.L.; Johnsen-Soriano, S.; Romero, F.J. Oxidative stress in keratoconus? Invest. Ophthalmol. Vis. Sci 2011, 52, 8592–8597. [Google Scholar]
- Uchida, K. Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med 2000, 28, 1685–1696. [Google Scholar]
- Kalra, J.; Mantha, S.V.; Kumar, P.; Prasad, K. Protective effects of lazaroids against oxygen-free radicals induced lysosomal damage. Mol. Cell. Biochem 1994, 136, 125–129. [Google Scholar]
- Behndig, A.; Karlsson, K.; Johansson, B.O.; Brännström, T.; Marklund, S.L. Superoxide dismutase isoenzymes in the normal and diseased human cornea. Invest. Ophthalmol. Vis. Sci 2001, 42, 2293–2296. [Google Scholar]
- Brown, D.J.; Lin, B.; Chwa, M.; Atilano, S.R.; Kim, D.W.; Kenney, M.C. Elements of the nitric oxide pathway can degrade TIMP-1 and increase gelatinase activity. Mol. Vis 2004, 10, 281–288. [Google Scholar]
- Olofsson, E.M.; Marklund, S.L.; Pedrosa-Domellöf, F.; Behndig, A. Interleukin-1alpha downregulates extracellular-superoxide dismutase in human corneal keratoconus stromal cells. Mol. Vis 2007, 13, 1285–1290. [Google Scholar]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med 2002, 33, 337–349. [Google Scholar]
- Pappa, A.; Estey, T.; Manzer, R.; Brown, D.; Vasiliou, V. Human aldehyde dehydrogenase 3A1 (ALDH3A1): Biochemical characterization and immunohistochemical localization in the cornea. Biochem. J 2003, 376, 615–23. [Google Scholar]
- Owen, J.B.; Butterfield, D.A. Measurement of oxidized/reduced glutathione ratio. Methods Mol. Biol 2010, 648, 269–277. [Google Scholar]
- Collier, S.A. Is the corneal degradation in keratoconus caused by matrix-metalloproteinases? Clin. Exp. Ophthalmol 2001, 29, 340–344. [Google Scholar]
- Guedez, L.; Stetler-Stevenson, W.G.; Wolff, L.; Wang, J.; Fukushima, P.; Mansoor, A.; Stetler-Stevenson, M. In vitro suppression of programmed cell death of B cells by tissue inhibitor of metalloproteinases-1. J. Clin. Invest 1998, 102, 2002–2010. [Google Scholar]
- Han, X.; Sun, Y.; Scott, S.; Bleich, D. Tissue inhibitor of metalloproteinase-1 prevents cytokine-mediated dysfunction and cytotoxicity in pancreatic islets and beta-cells. Diabetes 2001, 50, 1047–1055. [Google Scholar]
- Smith, V.A.; Hoh, H.B.; Littleton, M.; Easty, D.L. Over-expression of a gelatinase A activity in keratoconus. Eye 1995, 9, 429–433. [Google Scholar]
- Zhou, L.; Sawaguchi, S.; Twining, S.S.; Sugar, J.; Feder, R.S.; Yue, B.Y. Expression of degradative enzymes and protease inhibitors in corneas with keratoconus. Invest. Ophthalmol. Vis. Sci 1998, 39, 1117–1124. [Google Scholar]
- Atilano, S.R.; Coskun, P.; Chwa, M.; Jordan, N.; Reddy, V.; Le, K.; Wallace, D.C.; Kenney, M.C. Accumulation of mitochondrial DNA damage in keratoconus corneas. Invest. Ophthalmol. Vis. Sci 2005, 46, 1256–1263. [Google Scholar]
- De Grey, A.D.N.J. An Introduction to Mitochondria. History of the Mitochondrial Free Radical Theory of Aging, 1954–1995. In The Mitochondrial Free Radical Theory of Aging; R.G. Landes Company: Austin, TX, USA, 1999. [Google Scholar]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal. Transduct 2012, 2012, 646354. [Google Scholar]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet 2005, 6, 389–402. [Google Scholar]
- Eghrari, A.O.; Gottsch, J.D. Fuchs’ corneal dystrophy. Expert Rev. Ophthalmol 2010, 5, 147–159. [Google Scholar]
- Thalamuthu, A.; Khor, C.C.; Venkataraman, D.; Koh, L.W.; Tan, D.T.; Aung, T.; Mehta, J.S.; Vithana, E.N. Association of TCF4 gene polymorphisms with Fuchs’ corneal dystrophy in the Chinese. Invest. Ophthalmol. Vis. Sci 2011, 52, 5573–5578. [Google Scholar]
- Klintworth, G.K. Corneal dystrophies. Orphanet J. Rare Dis 2009, 4, 7. [Google Scholar]
- Schrems-Hoesl, L.M.; Schrems, W.A.; Cruzat, A.; Shahatit, B.M.; Bayhan, H.A.; Jurkunas, U.V.; Hamrah, P. Cellular and subbasal nerve alterations in early stage Fuchs’ endothelial corneal dystrophy: An in vivo confocal microscopy study. Eye 2013, 27, 42–49. [Google Scholar]
- Elhalis, H.; Azizi, B.; Jurkunas, U.V. Fuchs endothelial corneal dystrophy. Ocul. Surf 2010, 8, 173–184. [Google Scholar]
- Schmedt, T.; Silva, M.M.; Ziaei, A.; Jurkunas, U. Molecular bases of corneal endothelial dystrophies. Exp. Eye Res 2012, 95, 24–34. [Google Scholar]
- Peh, G.S.; Beuerman, R.W.; Colman, A.; Tan, D.T.; Mehta, J.S. Human corneal endothelial cell expansion for corneal endothelium transplantation: An overview. Transplantation 2011, 91, 811–819. [Google Scholar]
- Bonanno, J.A. Identity and regulation of ion transport mechanisms in the corneal endothelium. Prog. Retin. Eye Res 2003, 22, 69–94. [Google Scholar]
- Iliff, B.W.; Riazuddin, S.A.; Gottsch, J.D. The genetics of Fuchs’ corneal dystrophy. Expert Rev. Ophthalmol 2012, 7, 363–375. [Google Scholar]
- Hemadevi, B.; Srinivasan, M.; Arunkumar, J.; Prajna, N.V.; Sundaresan, P. Genetic analysis of patients with Fuchs endothelial corneal dystrophy in India. BMC Ophthalmol 2010, 10, 3. [Google Scholar]
- Riazuddin, S.A.; Parker, D.S.; McGlumphy, E.J.; Oh, E.C.; Iliff, B.W.; Schmedt, T.; Jurkunas, U.; Schleif, R.; Katsanis, N.; Gottsch, J.D. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am. J. Hum. Genet 2012, 90, 533–953. [Google Scholar]
- Gottsch, J.D.; Bowers, A.L.; Margulies, E.H.; Seitzman, G.D.; Kim, S.W.; Saha, S.; Jun, A.S.; Stark, W.J.; Liu, S.H. Serial analysis of gene expression in the corneal endothelium of Fuchs’ dystrophy. Invest. Ophthalmol. Vis. Sci 2003, 44, 594–599. [Google Scholar]
- Jurkunas, U.V.; Rawe, I.; Bitar, M.S.; Zhu, C.; Harris, D.L.; Colby, K.; Joyce, N.C. Decreased expression of peroxiredoxins in Fuchs’ endothelial dystrophy. Invest. Ophthalmol. Vis. Sci 2008, 49, 2956–2963. [Google Scholar]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst 2000, 92, 1564–1572. [Google Scholar]
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res 2010, 12, R44. [Google Scholar]
- Nordberg, J.; Arnér, E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med 2001, 31, 1287–1312. [Google Scholar]
- Carlson, B.A.; Yoo, M.H.; Tobe, R.; Mueller, C.; Naranjo-Suarez, S.; Hoffmann, V.J.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin reductase 1 protects against chemically induced hepatocarcinogenesis via control of cellular redox homeostasis. Carcinogenesis 2012, 33, 1806–1813. [Google Scholar]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem 2003, 278, 12029–12038. [Google Scholar]
- Ishii, T.; Itoh, K.; Takahashi, S.; Sato, H.; Yanagawa, T.; Katoh, Y.; Bannai, S.; Yamamoto, M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J. Biol. Chem 2000, 275, 16023–16029. [Google Scholar]
- Czarny, P.; Kasprzak, E.; Wielgorski, M.; Udziela, M.; Markiewicz, B.; Blasiak, J.; Szaflik, J.; Szaflik, J.P. DNA damage and repair in Fuchs endothelial corneal dystrophy. Mol. Biol. Rep 2013, 40, 2977–2983. [Google Scholar]
- Gredilla, R. DNA damage and base excision repair in mitochondria and their role in aging. J. Aging Res 2010, 2011, 257093. [Google Scholar]
- Kirkinezos, I.G.; Moraes, C.T. Reactive oxygen species and mitochondrial diseases. Semin. Cell Dev. Biol 2001, 12, 449–457. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wojcik, K.A.; Kaminska, A.; Blasiak, J.; Szaflik, J.; Szaflik, J.P. Oxidative Stress in the Pathogenesis of Keratoconus and Fuchs Endothelial Corneal Dystrophy. Int. J. Mol. Sci. 2013, 14, 19294-19308. https://doi.org/10.3390/ijms140919294
Wojcik KA, Kaminska A, Blasiak J, Szaflik J, Szaflik JP. Oxidative Stress in the Pathogenesis of Keratoconus and Fuchs Endothelial Corneal Dystrophy. International Journal of Molecular Sciences. 2013; 14(9):19294-19308. https://doi.org/10.3390/ijms140919294
Chicago/Turabian StyleWojcik, Katarzyna A., Anna Kaminska, Janusz Blasiak, Jerzy Szaflik, and Jacek P. Szaflik. 2013. "Oxidative Stress in the Pathogenesis of Keratoconus and Fuchs Endothelial Corneal Dystrophy" International Journal of Molecular Sciences 14, no. 9: 19294-19308. https://doi.org/10.3390/ijms140919294