Mesoscale Characterization of Supramolecular Transient Networks Using SAXS and Rheology


 ,
,
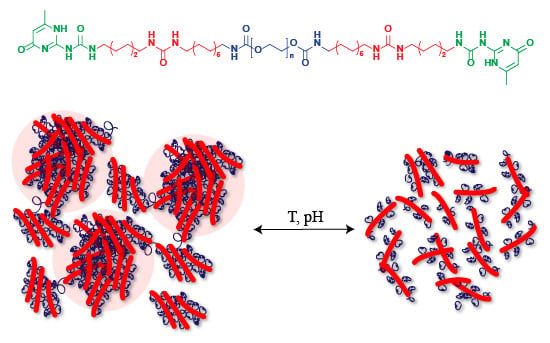
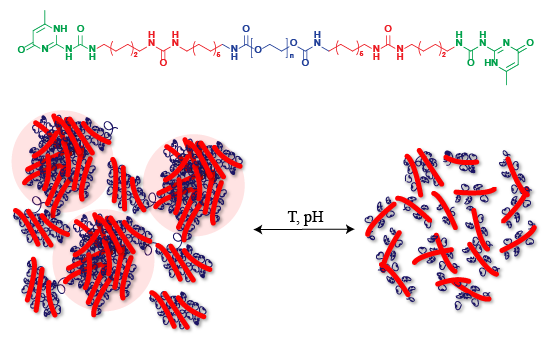
Abstract
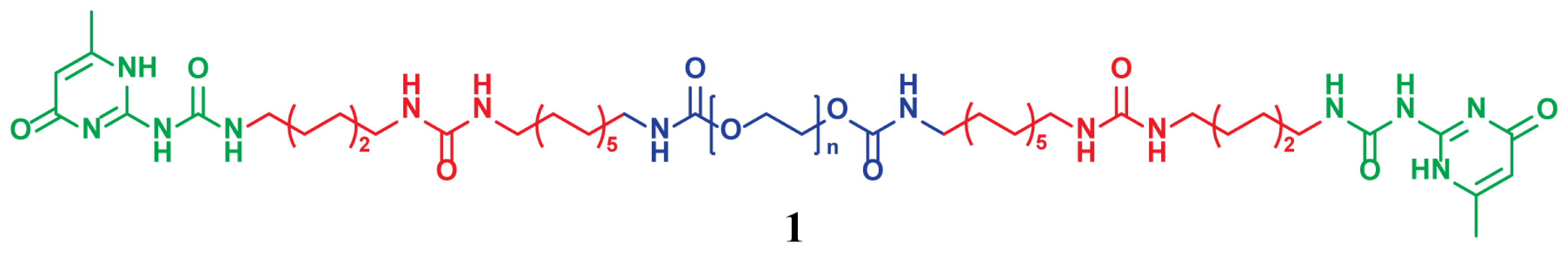
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
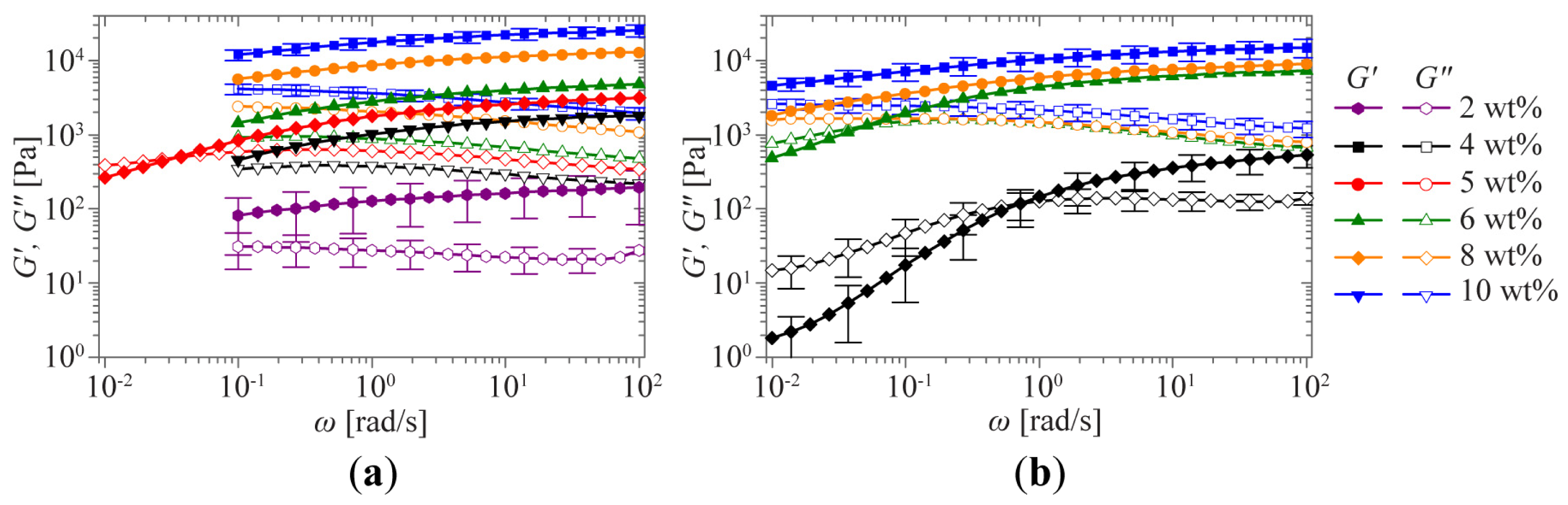
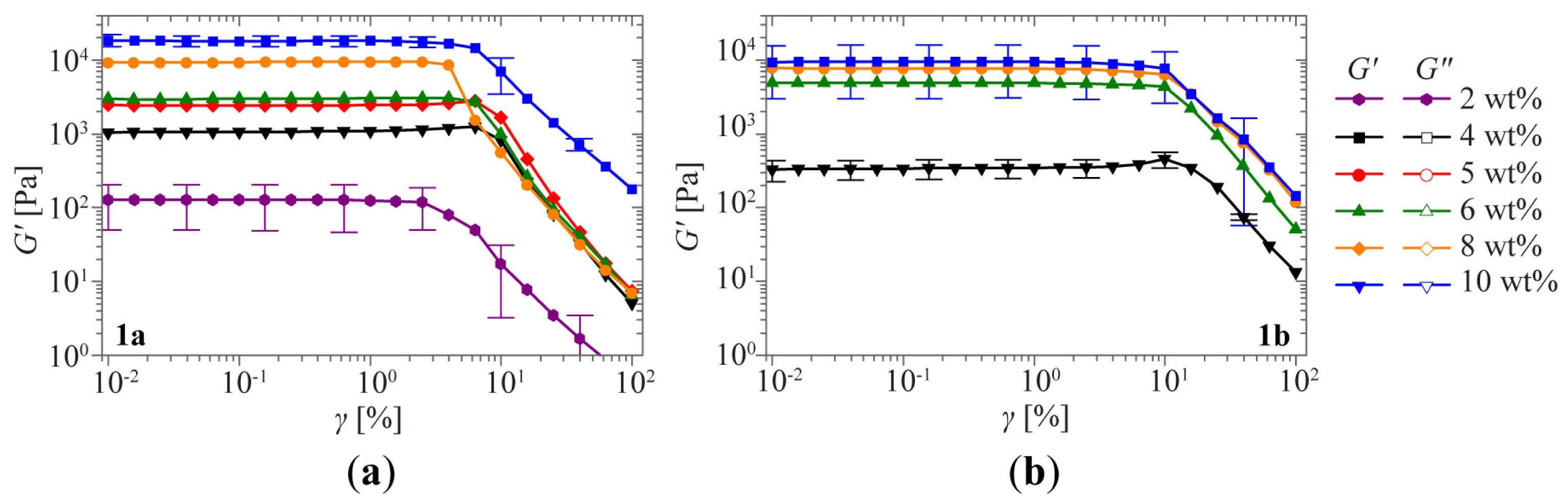
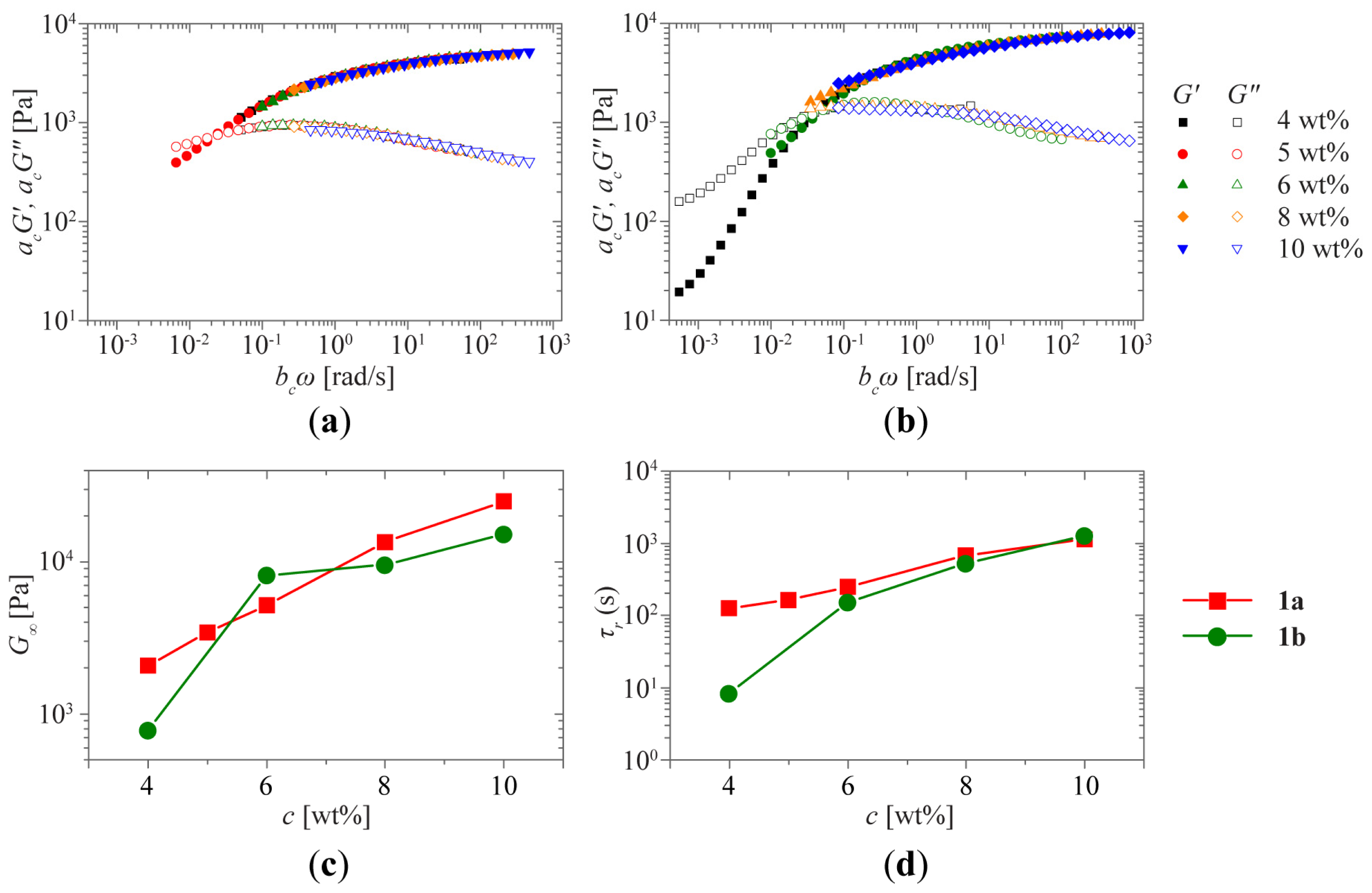
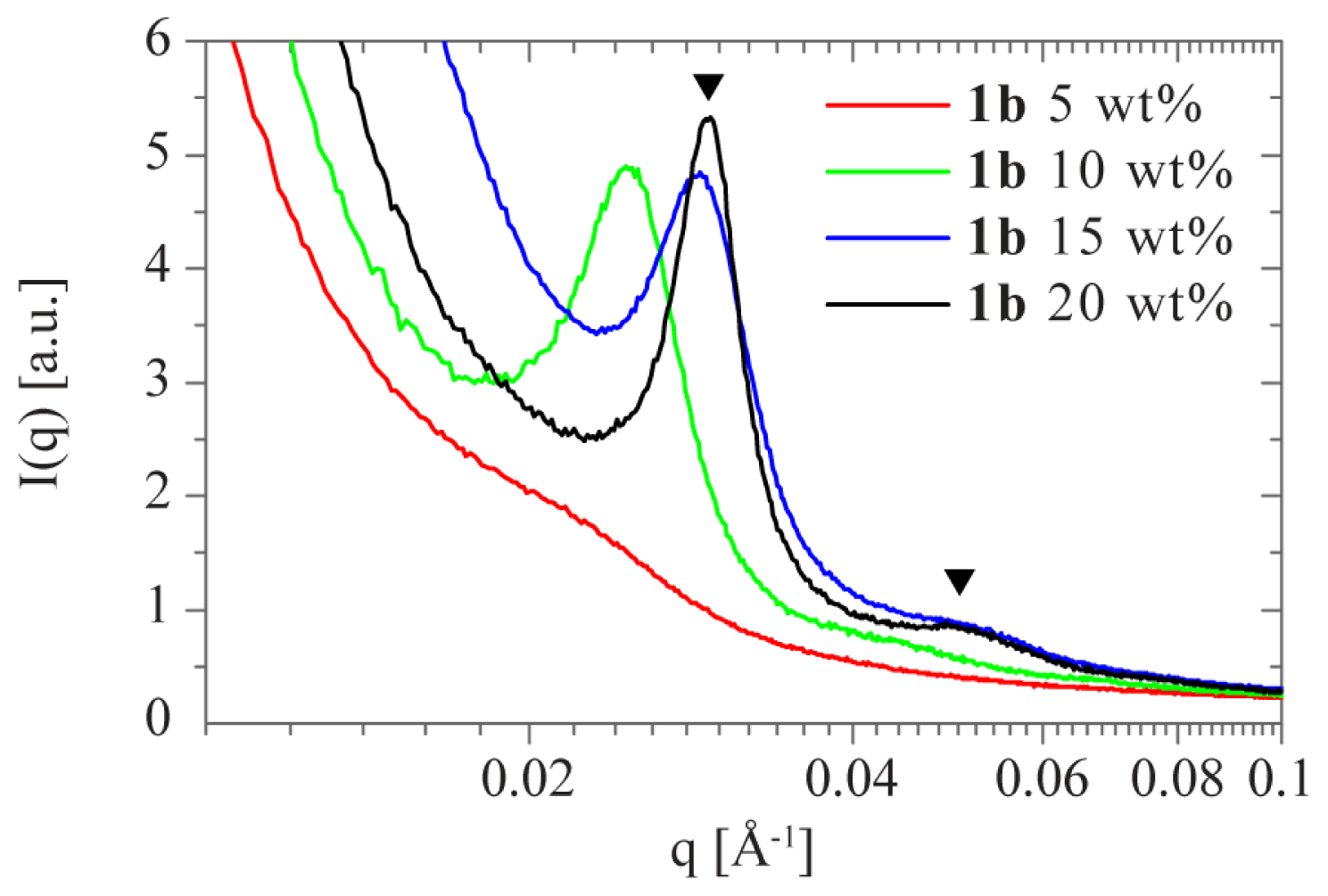
2.1. Sol-Gel Transition Triggered by Concentration
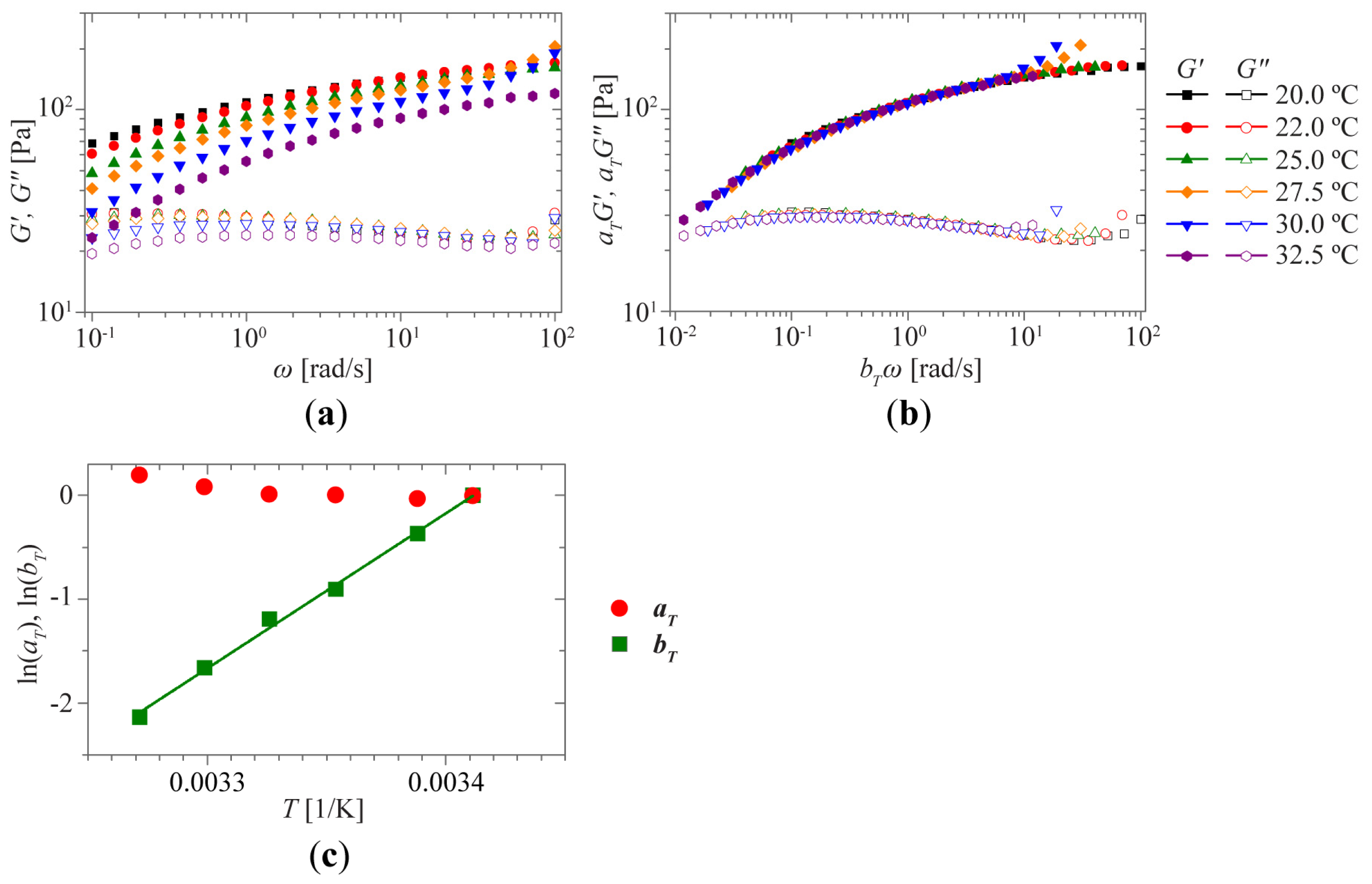
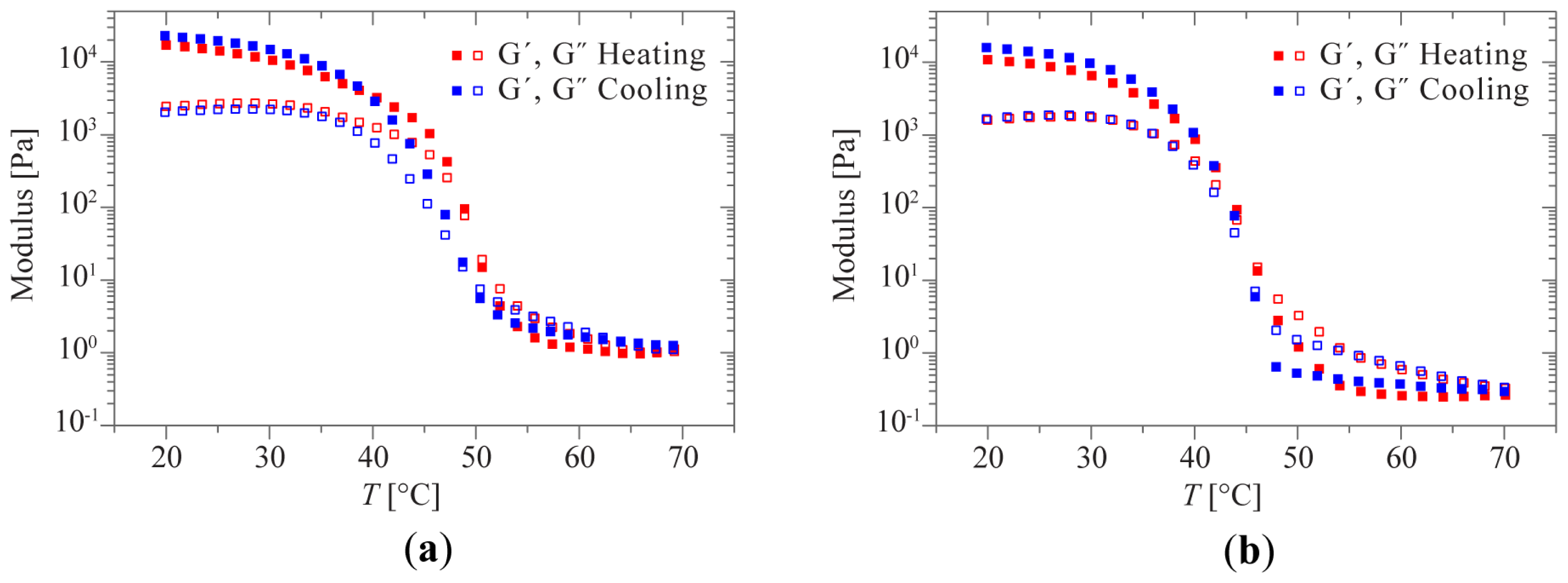
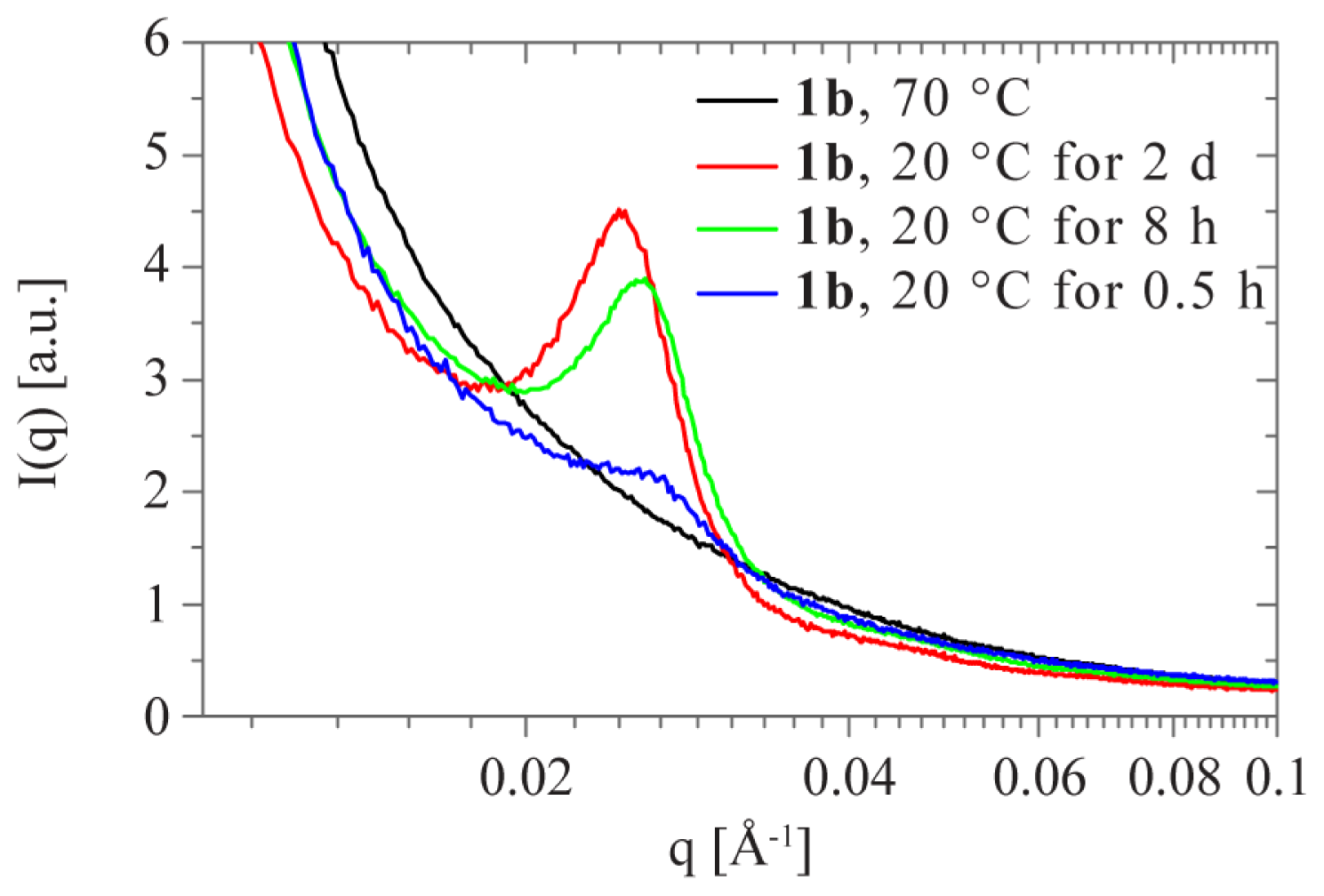
2.2. Sol-Gel Transition Triggered by Temperature
2.3. Sol-Gel Transition Triggered by pH
3. Experimental Section
3.1. Materials
3.2. Methods
3.2.1. Rheology
3.2.2. Small-Angle X-ray Scattering
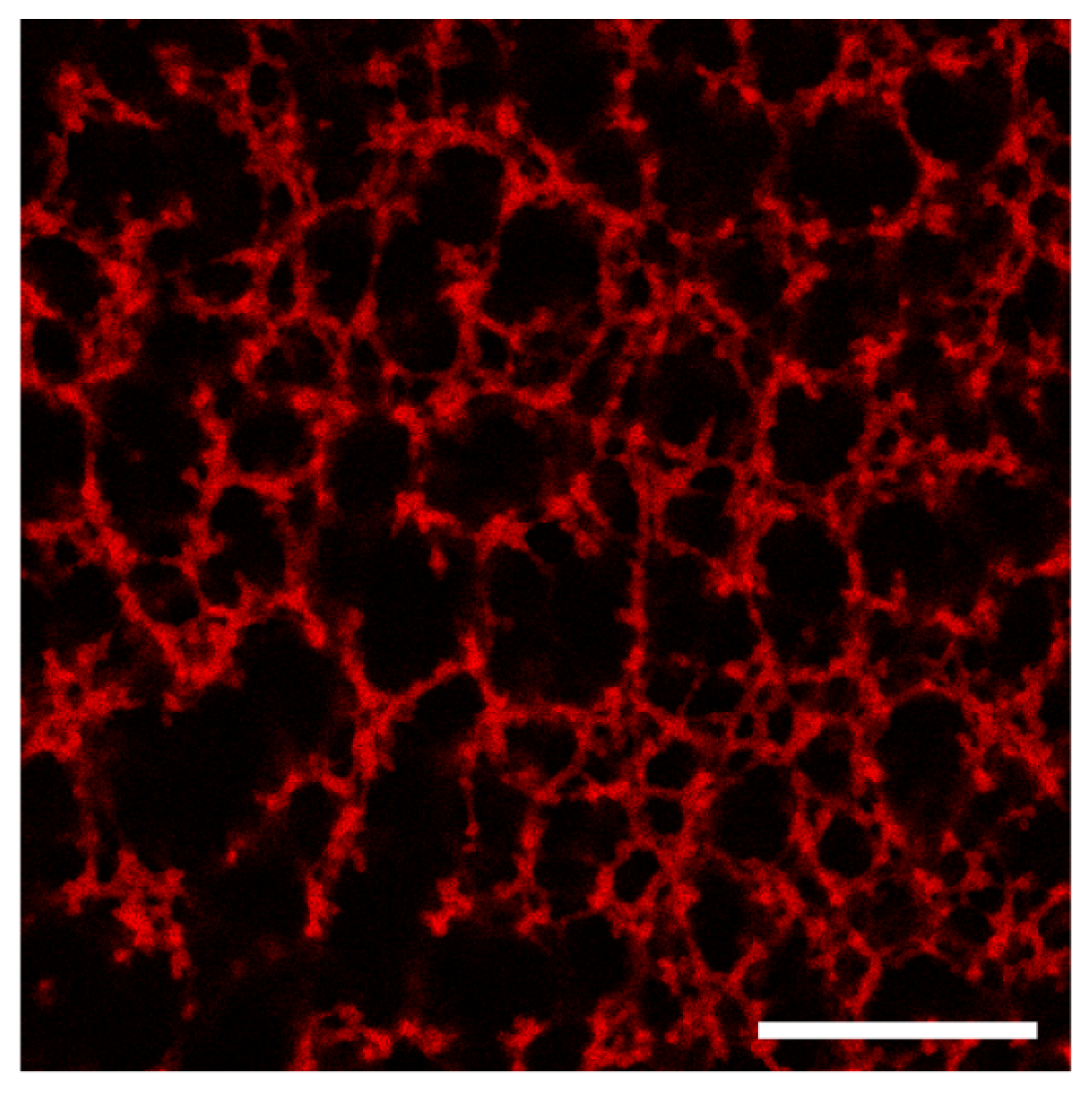
3.2.3. Confocal Microscopy
4. Conclusions
Supplementary Information
ijms-15-01096-s001.pdfAcknowledgments
Conflicts of Interest
References
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater 2006, 18, 1345–1360. [Google Scholar]
- Lutolf, M.P.; Hubbell, J.A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol 2005, 23, 47–55. [Google Scholar]
- Kloxin, A.M.; Kasko, A.M.; Salinas, C.N.; Anseth, K.S. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science 2009, 324, 59–63. [Google Scholar]
- Aida, T.; Meijer, E.W.; Stupp, S.I. Functional supramolecular polymers. Science 2012, 335, 813–817. [Google Scholar]
- Wojtecki, R.J.; Meador, M.A.; Rowan, S.J. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater 2011, 10, 14–27. [Google Scholar]
- Berret, J.-F.; Calvet, D.; Collet, A.; Viguier, M. Fluorocarbon associative polymers. Curr. Opin. Colloid Interface Sci 2003, 8, 296–306. [Google Scholar]
- Chassenieux, C.; Nicolai, T.; Benyahia, L. Rheology of associative polymer solutions. Curr. Opin. Colloid Interface Sci 2011, 16, 18–26. [Google Scholar]
- Brunsveld, L.; Folmer, B.J.B.; Meijer, E.W.; Sijbesma, R.P. Supramolecular polymers. Chem. Rev 2001, 101, 4071–4098. [Google Scholar]
- Pawar, G.M.; Koenigs, M.; Fahimi, Z.; Cox, M.; Voets, I.K.; Wyss, H.M.; Sijbesma, R.P. Injectable hydrogels from segmented PEG-bisurea copolymers. Biomacromolecules 2012, 13, 3966–3976. [Google Scholar]
- Leenders, C.M.A.; Albertazzi, L.; Mes, T.; Koenigs, M.M.E.; Palmans, A.R.A.; Meijer, E.W. Supramolecular polymerization in water harnessing both hydrophobic effects and hydrogen bond formation. Chem. Commun 2013, 49, 1963–1965. [Google Scholar]
- Capito, R.M.; Azevedo, H.S.; Velichko, Y.S.; Mata, A.; Stupp, S.I. Self-assembly of large and small molecules into hierarchically ordered sacs and membranes. Science 2008, 319, 1812–1816. [Google Scholar]
- Van Bommel, K.J.C.; van der Pol, C.; Muizebelt, I.; Friggeri, A.; Heeres, A.; Meetsma, A.; Feringa, B.L.; van Esch, J. Responsive cyclohexane-based low-molecular-weight hydrogelators with modular architecture. Angew. Chem. Int. Ed 2004, 43, 1663–1667. [Google Scholar]
- Krieg, E.; Rybtchinski, B. Noncovalent water-based materials: Robust yet adaptive. Chem. Eur. J 2011, 17, 9016–9026. [Google Scholar]
- Kouwer, P.H.J.; Koepf, M.; le Sage, V.A.A.; Jaspers, M.; van Buul, A.M.; Eksteen-Akeroyd, Z.H.; Woltinge, T.; Schwartz, E.; Kitto, H.J.; Hoogenboom, R.; et al. Responsive biomimetic networks from polyisocyanopeptide hydrogels. Nature 2013. [Google Scholar] [CrossRef]
- Schappacher, M.; Deffieux, A.; Meins, J.-F.L. Soft dynamic covalent hydrogels based on iron(III)tetraphenylporphyrinato-functionalized 4-arm poly(ethylene oxide). Polym. Chem 2013, 4, 458–461. [Google Scholar]
- Kieltyka, R.E.; Pape, A.C.H.; Albertazzi, L.; Nakano, Y.; Bastings, M.M.C.; Voets, I.K.; Dankers, P.Y.W.; Meijer, E.W. Mesoscale modulation of supramolecular ureidopyrimidinone-based Poly(ethylene glycol) transient networks in water. J. Am. Chem. Soc 2013, 135, 11159–11164. [Google Scholar]
- Fenske, T.; Korth, H.-G.; Mohr, A.; Schmuck, C. Advances in switchable supramolecular nanoassemblies. Chem. Eur. J 2012, 18, 738–755. [Google Scholar]
- Bastings, M.M.C.; Koudstaal, S.; Kieltyka, R.E.; Nakano, Y.; Pape, A.C.H.; Feyen, D.A.M.; van Slochteren, F.J.; Doevendans, P.A.; Sluijter, J.P.G.; Meijer, E.W.; et al. A fast pH-switchable and self-healing supramolecular hydrogel carrier for guided, local catheter injection in the infarcted myocardium. Adv. Healthc. Mater 2014, 3, 70–78. [Google Scholar]
- Chen, Q.; Zhu, L.; Zhao, C.; Wang, Q.; Zheng, J. A robust, one-pot synthesis of highly mechanical and recoverable double network hydrogels using thermoreversible sol-gel polysaccharide. Adv. Mater 2013, 25, 4171–4176. [Google Scholar]
- Gong, J.P.; Osada, Y. Soft and Wet Materials: From Hydrogels to Biotissues. In High Solid Dispersions; Cloitre, M., Ed.; Advances in Polymer Science, Springer: Berlin/Heidelberg, Germany, 2010; Volume 236, pp. 203–246. [Google Scholar]
- Guo, H.; Zhang, J.; Xu, T.; Zhang, Z.; Yao, J.; Shao, Z. The robust hydrogel hierarchically assembled from a pH sensitive peptide amphiphile based on silk fibroin. Biomacromolecules 2013, 14, 2733–2738. [Google Scholar]
- Lemmers, M.; Voets, I.K.; Stuart, M.A.C.; van der Gucht, J. Transient network topology of interconnected polyelectrolyte complex micelles. Soft Matter 2011, 7, 1378–1389. [Google Scholar]
- Miasnikova, A.; Laschewsky, A.; de Paoli, G.; Papadakis, C.M.; Müller-Buschbaum, P.; Funari, S.S. Thermoresponsive hydrogels from symmetrical triblock copolymers poly(styrene-block-(methoxy diethylene glycol acrylate)-block-styrene). Langmuir 2012, 28, 4479–4490. [Google Scholar]
- Krogstad, D.V.; Lynd, N.A.; Choi, S.-H.; Spruell, J.M.; Hawker, C.J.; Kramer, E.J.; Tirrell, M.V. Effects of polymer and salt concentration on the structure and properties of triblock copolymer coacervate hydrogels. Macromolecules 2013, 46, 1512–1518. [Google Scholar]
- Dankers, P.Y.W.; Hermans, T.M.; Baughman, T.W.; Kamikawa, Y.; Kieltyka, R.E.; Bastings, M.M.C.; Janssen, H.M.; Sommerdijk, N.A.J.M.; Larsen, A.; van Luyn, M.J.; et al. Hierarchical formation of supramolecular transient networks in water: A modular injectable delivery system. Adv. Mater 2012, 24, 2703–2709. [Google Scholar]
- Dankers, P.Y.W.; van Luyn, M.J.A.; Huizinga-van der Vlag, A.; van Gemert, G.M.L.; Petersen, A.H.; Meijer, E.W.; Janssen, H.M.; Bosman, A.W.; Popa, E.R. Development and in-vivo characterization of supramolecular hydrogels for intrarenal drug delivery. Biomaterials 2012, 33, 5144–5155. [Google Scholar]
- Guo, M.; Cao, X.; Meijer, E.W.; Dankers, P.Y.W. Core-shell capsules based on supramolecular hydrogels show shell-related erosion and release due to confinement. Macromol. Biosci 2013, 13, 77–83. [Google Scholar]
- Weiss, R.G.; Terech, P. Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Springer: Dordrecht, The Netherlands, 2006. [Google Scholar]
- Seiffert, S.; Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev 2012, 41, 909–930. [Google Scholar]
- Annable, T.; Buscall, R.; Ettelaie, R.; Whittlestone, D. The rheology of solutions of associating polymers: Comparison of experimental behavior with transient network theory. J. Rheol 1993, 37, 695–726. [Google Scholar]
- Meins, J.-F.L.; Tassin, J.-F. Elastic modulus and relaxation times in telechelic associating polymers. Colloid Polym. Sci 2003, 281, 283–287. [Google Scholar]
- MacKintosh, F.C.; Käs, J.; Janmey, P.A. Elasticity of semiflexible biopolymer networks. Phys. Rev. Lett 1995, 75, 4425–4428. [Google Scholar]
- Lin, Y.-C.; Yao, N.Y.; Broedersz, C.P.; Herrmann, H.; MacKintosh, F.C.; Weitz, D.A. Origins of Elasticity in Intermediate Filament Networks. Phys. Rev. Lett 2010, 104, 058101. [Google Scholar]
- Gardel, M.L.; Shin, J.H.; MacKintosh, F.C.; Mahadevan, L.; Matsudaira, P.; Weitz, D.A. Elastic behavior of cross-linked and bundled actin networks. Science 2004, 304, 1301–1305. [Google Scholar]
- Semenov, A.N.; Rubinstein, M. Thermoreversible gelation in solutions of associative polymers. 1. Statics. Macromolecules 1998, 31, 1373–1385. [Google Scholar]
- Rubinstein, M.; Semenov, A.N. Thermoreversible gelation in solutions of associating polymers. 2. Linear Dynamics. Macromolecules 1998, 31, 1386–1397. [Google Scholar]
- Hackelbusch, S.; Rossow, T.; van Assenbergh, P.; Seiffert, S. Chain dynamics in supramolecular polymer networks. Macromolecules 2013, 46, 6273–6286. [Google Scholar]
- Mendes, E., Jr.; Lindner, P.; Buzier, M.; Boué, F.; Bastide, J. Experimental evidence for inhomogeneous swelling and deformation in statistical gels. Phys. Rev. Lett 1991, 66, 1595–1598. [Google Scholar]
- Waters, D.J.; Engberg, K.; Parke-Houben, R.; Hartmann, L.; Ta, C.N.; Toney, M.F.; Frank, C.W. Morphology of photopolymerized end-linked poly(ethylene glycol) hydrogels by small-angle X-ray scattering. Macromolecules 2010, 43, 6861–6870. [Google Scholar]
- Seitz, M.E.; Burghardt, W.R.; Shull, K.R. Micelle morphology and mechanical response of triblock gels. Macromolecules 2009, 42, 9133–9140. [Google Scholar]











© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pape, A.C.H.; Bastings, M.M.C.; Kieltyka, R.E.; Wyss, H.M.; Voets, I.K.; Meijer, E.W.; Dankers, P.Y.W. Mesoscale Characterization of Supramolecular Transient Networks Using SAXS and Rheology. Int. J. Mol. Sci. 2014, 15, 1096-1111. https://doi.org/10.3390/ijms15011096
Pape ACH, Bastings MMC, Kieltyka RE, Wyss HM, Voets IK, Meijer EW, Dankers PYW. Mesoscale Characterization of Supramolecular Transient Networks Using SAXS and Rheology. International Journal of Molecular Sciences. 2014; 15(1):1096-1111. https://doi.org/10.3390/ijms15011096
Chicago/Turabian StylePape, A. C. H., Maartje M. C. Bastings, Roxanne E. Kieltyka, Hans M. Wyss, Ilja K. Voets, E. W. Meijer, and Patricia Y. W. Dankers. 2014. "Mesoscale Characterization of Supramolecular Transient Networks Using SAXS and Rheology" International Journal of Molecular Sciences 15, no. 1: 1096-1111. https://doi.org/10.3390/ijms15011096