Regulation of TRAIL-Receptor Expression by the Ubiquitin-Proteasome System

Abstract
:
1. Introduction
2. Modulation of Tumor Necrosis Factor (TNF)-Related Apoptosis-Inducing Ligand (TRAIL)-Receptors in Cancer
2.1. TRAIL-Receptor Expression in Healthy vs. Malignant Tissue
2.2. TRAIL-Ligand vs. Fas Ligand (FasL)
2.3. TRAIL-Induced Apoptosis Pathways

2.4. TRAIL Targeted Therapies
2.5. Resistance to TRAIL-Targeted Therapies
2.6. The Role of Ubiquitin in Regulating TRAIL Receptors

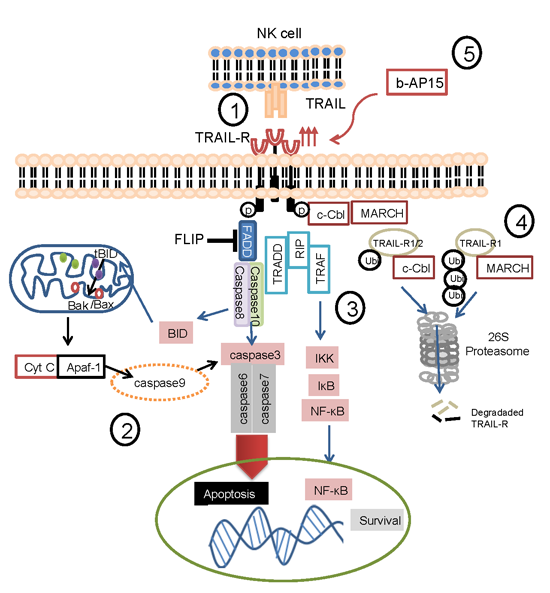{kind=link}
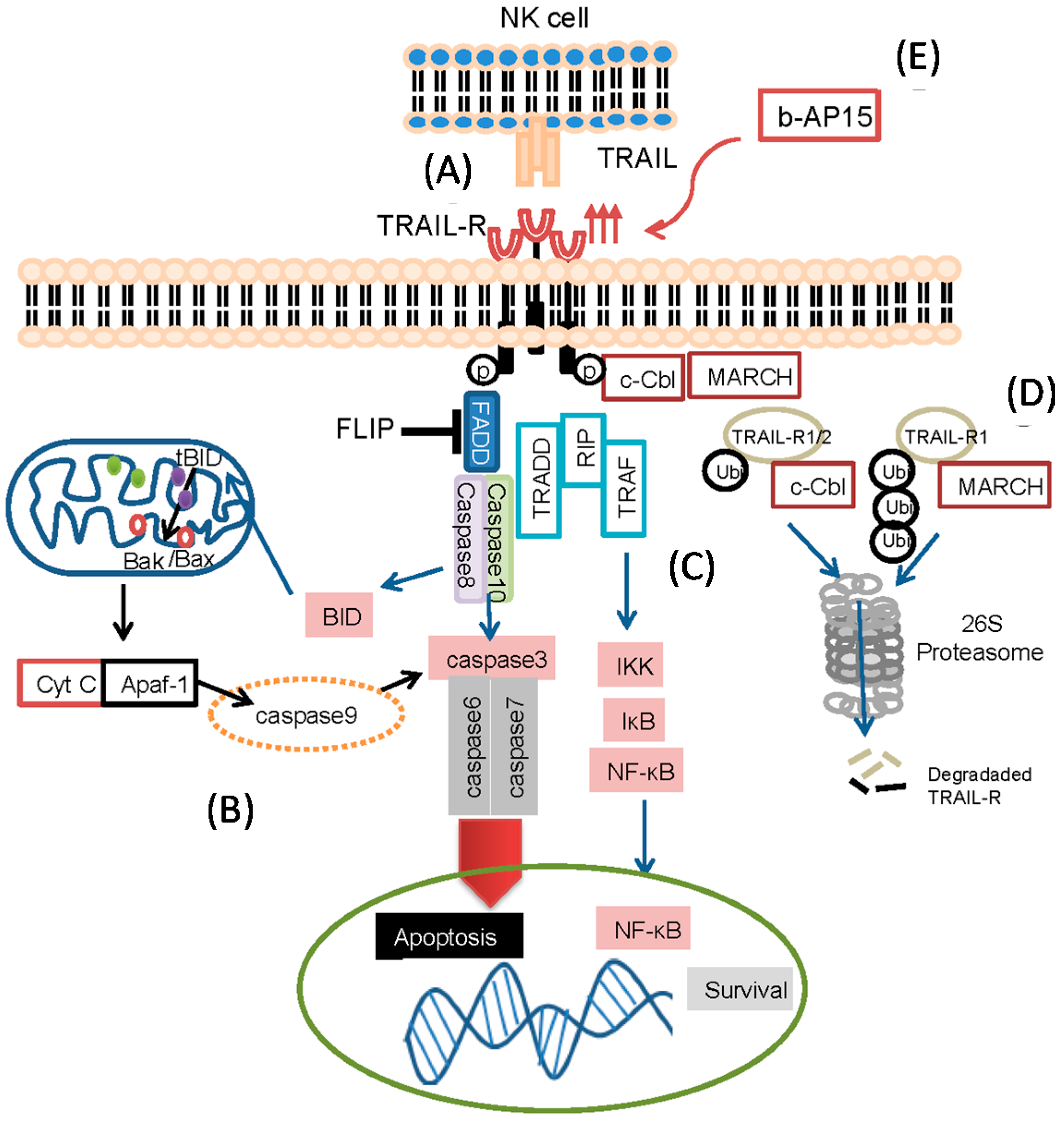{kind=link}
| Ligase | Target | Outcome |
|---|---|---|
| c-Cbl | TRAIL-R1 and R2 | Mono-ubiquitination and degradation of internalized receptors by the proteasome |
| MARCH-1, -2, and -9 | TRAIL-R1 and R2 | Poly-ubiquitination, internalization and degradation of internalized receptors by the lysosomal pathway |
| A20 | Procaspase-8 | K63-linked ubiquitination of RIP1 and down-regulation of caspase-8 activity |
| HECTD3 | Caspase-8 | Facilitates survival by promoting K63-linked polyubiquitination of caspase-8 |
| Itch | FLIP | Target FLIP for proteasomal degradation |
| CUL3 | Procaspase-8 | Ubiqutination of procaspase-8 to create binding sites for the scaffold protein p62/SQSTM |
2.7. Enhancing TRAIL-Mediated Apoptosis by Targeting Ubiquitin-Proteasome System (UPS)
3. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740C, 364–378. [Google Scholar] [CrossRef]
- Watts, T.H. TNF/TNFR family members in costimulation of T cell responses. Ann. Rev. Immunol 2005, 23, 23–68. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Nesterov, A.; Nikrad, M.; Johnson, T.; Kraft, A.S. Oncogenic Ras sensitizes normal human cells to tumor necrosis factor-α-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2004, 64, 3922–3927. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, D.W.; Shah, K. TRAIL on trial: Preclinical advances in cancer therapy. Trends Mol. Med. 2013, 19, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Wennerberg, E.; D’Arcy, P.; Gurajada, D.; Linder, S.; Lundqvist, A. A novel inhibitor of proteasome deubiquitinating activity renders tumor cells sensitive to TRAIL-mediated apoptosis by natural killer cells and T cells. Cancer Immunol. Immunother. 2013, 62, 1359–1368. [Google Scholar] [CrossRef]
- Dokouhaki, P.; Schuh, N.W.; Joe, B.; Allen, C.A.; Der, S.D.; Tsao, M.S.; Zhang, L. NKG2D regulates production of soluble TRAIL by ex vivo expanded human γδ T cells. Eur. J. Immunol. 2013, 43, 3175–3182. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Olson, D.; Tardivel, A.; Browning, B.; Lugovskoy, A.; Gong, D.; Dobles, M.; Hertig, S.; Hofmann, K.; van Vlijmen, H.; et al. Identification of a new murine tumor necrosis factor receptor locus that contains two novel murine receptors for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Biol. Chem. 2003, 278, 5444–5454. [Google Scholar] [CrossRef]
- Shirley, S.; Morizot, A.; Micheau, O. Regulating TRAIL receptor-induced cell death at the membrane: A deadly discussion. Recent Pat. Anti-Cancer Drug Dis. 2011, 6, 311–323. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Maecker, H.; Sharp, D.; Lawrence, D.; Renz, M.; Vucic, D.; Ashkenazi, A. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 2005, 280, 40599–40608. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.A.; Turley, H.; Kimberley, F.C.; Liu, X.S.; Mongkolsapaya, J.; Ch’En, P.; Xu, X.N.; Jin, B.Q.; Pezzella, F.; Screaton, G.R. Expression of TRAIL and TRAIL receptors in normal and malignant tissues. Cell Res. 2005, 15, 430–438. [Google Scholar] [CrossRef]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol. 2002, 168, 1356–1361. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; DiVito, K.A.; Sznol, M.; Kovacs, D.; Halaban, R.; Berger, A.J.; Flaherty, K.T.; Camp, R.L.; Lazova, R.; Rimm, D.L.; et al. Expression of tumor necrosis factor—Related apoptosis-inducing ligand receptors 1 and 2 in melanoma. Clin. Cancer Res. 2006, 12, 3856–3863. [Google Scholar] [CrossRef]
- Perraud, A.; Akil, H.; Nouaille, M.; Petit, D.; Labrousse, F.; Jauberteau, M.O.; Mathonnet, M. Expression of p53 and DR5 in normal and malignant tissues of colorectal cancer: Correlation with advanced stages. Oncol. Rep. 2011, 26, 1091–1097. [Google Scholar] [PubMed]
- Gallmeier, E.; Bader, D.C.; Kriegl, L.; Berezowska, S.; Seeliger, H.; Goke, B.; Kirchner, T.; Bruns, C.; de Toni, E.N. Loss of TRAIL-receptors is a recurrent feature in pancreatic cancer and determines the prognosis of patients with no nodal metastasis after surgery. PLoS One 2013, 8, e56760. [Google Scholar] [CrossRef] [PubMed]
- Sanlioglu, A.D.; Korcum, A.F.; Pestereli, E.; Erdogan, G.; Karaveli, S.; Savas, B.; Griffith, T.S.; Sanlioglu, S. TRAIL death receptor-4 expression positively correlates with the tumor grade in breast cancer patients with invasive ductal carcinoma. Int J Radiat. Oncol. Biol. Phys. 2007, 69, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Ruffin, N.; Ahmed, S.S.; Osorio, L.M.; Wang, X.N.; Jackson, G.H.; Collin, M.P.; Ekre, H.P.; Chiodi, F.; Dickinson, A.M. The involvement of epithelial Fas in a human model of graft versus host disease. Transplantation 2011, 91, 946–951. [Google Scholar] [CrossRef]
- Schungel, S.; Buitrago-Molina, L.E.; Nalapareddy, P.; Lebofsky, M.; Manns, M.P.; Jaeschke, H.; Gross, A.; Vogel, A. The strength of the Fas ligand signal determines whether hepatocytes act as type 1 or type 2 cells in murine livers. Hepatology 2009, 50, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Holoch, P.A.; Griffith, T.S. TNF-related apoptosis-inducing ligand (TRAIL): A new path to anti-cancer therapies. Eur. J. Pharmacol. 2009, 625, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.D.; Belz, G.T.; Fortner, K.A.; Budd, R.C.; Strasser, A.; Bouillet, P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity 2008, 28, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Weant, A.E.; Michalek, R.D.; Khan, I.U.; Holbrook, B.C.; Willingham, M.C.; Grayson, J.M. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity 2008, 28, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Griffiths, G.M. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat. Med. 1999, 5, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Kawasaki, A.; Ebata, T.; Ohmoto, H.; Ikeda, S.; Inoue, S.; Yoshino, K.; Okumura, K.; Yagita, H. Metalloproteinase-mediated release of human Fas ligand. J. Exp. Med. 1995, 182, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Itai, T.; Adachi, M.; Nagata, S. Down-regulation of Fas ligand by shedding. Nat. Med. 1998, 4, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, A.C.; Haux, J.; Steinkjer, B.; Nonstad, U.; Egeberg, K.; Sundan, A.; Ashkenazi, A.; Espevik, T. Regulation of Apo2 ligand/trail expression in NK cells-involvement in NK cell-mediated cytotoxicity. Cytokine 1999, 11, 664–672. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Takeda, K.; Akiba, H.; Tsutsui, H.; Okamura, H.; Nakanishi, K.; Okumura, K.; Yagita, H. Expression and function of TNF-related apoptosis-inducing ligand on murine activated NK cells. J. Immunol. 1999, 163, 1906–1913. [Google Scholar] [PubMed]
- Sato, K.; Hida, S.; Takayanagi, H.; Yokochi, T.; Kayagaki, N.; Takeda, K.; Yagita, H.; Okumura, K.; Tanaka, N.; Taniguchi, T.; et al. Antiviral response by natural killer cells through TRAIL gene induction by IFN-α/β. Eur. J. Immunol. 2001, 31, 3138–3146. [Google Scholar]
- Cummins, N.; Badley, A. The TRAIL to viral pathogenesis: The good, the bad and the ugly. Curr. Mol. Med. 2009, 9, 495–505. [Google Scholar] [CrossRef]
- Rethi, B.; Eidsmo, L. FasL and TRAIL signaling in the skin during cutaneous leishmaniasis—Implications for tissue immunopathology and infectious control. Front. Immunol. 2012, 3, 163. [Google Scholar] [CrossRef] [PubMed]
- Falschlehner, C.; Schaefer, U.; Walczak, H. Following TRAIL’s path in the immune system. Immunology 2009, 127, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Ueno, Y.; Ishii, M.; Yahagi, K.; Mano, Y.; Kisara, N.; Nakamura, N.; Shimosegawa, T.; Toyota, T.; Nagata, S. Fas-mediated cholangiopathy in the murine model of graft versus host disease. Hepatology 2000, 31, 966–974. [Google Scholar] [CrossRef]
- Sugimoto, M.; Shimaoka, M.; Hosotsubo, K.; Tanigami, H.; Taenaka, N.; Kiyono, H.; Yoshiya, I. Up-regulation of Fas ligand (FasL) mRNA expression in peripheral blood mononuclear cells (PBMC) after major surgery. Clin. Exp. Immunol. 1998, 112, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Murata, J.; Abe, R.; Shimizu, H. Increased soluble Fas ligand levels in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis preceding skin detachment. J. Allergy Clin. Immunol. 2008, 122, 992–1000. [Google Scholar] [CrossRef]
- Viard, I.; Wehrli, P.; Bullani, R.; Schneider, P.; Holler, N.; Salomon, D.; Hunziker, T.; Saurat, J.H.; Tschopp, J.; French, L.E. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998, 282, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Crissey, M.A.; Taub, R. Massive hepatic apoptosis associated with TGF-β1 activation after Fas ligand treatment of IGF binding protein-1-deficient mice. J. Clin. Investig. 2003, 111, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Sheard, M.A.; Asgharzadeh, S.; Liu, Y.; Lin, T.Y.; Wu, H.W.; Ji, L.; Groshen, S.; Lee, D.A.; Seeger, R.C. Membrane-bound TRAIL supplements natural killer cell cytotoxicity against neuroblastoma cells. J. Immunother. 2013, 36, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Knight, D.A.; Smyth, M.J.; Stewart, T.J. Sensitivity of a novel model of mammary cancer stem cell-like cells to TNF-related death pathways. Cancer Immunol. Immunother. 2012, 61, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, K.; Tanaka, Y.; Arihiro, K.; Kataoka, T.; Ohdan, H. Mechanistic analysis of the antitumor efficacy of human natural killer cells against breast cancer cells. Breast Cancer Res. Treat. 2012, 134, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Jazirehi, A.R.; Kurdistani, S.K.; Economou, J.S. Histone deacetylase inhibitor sensitizes apoptosis-resistant melanomas to cytotoxic human T lymphocytes through regulation of TRAIL/DR5 pathway. J. Immunol. 2014, 192, 3981–3989. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Dogan, Y.; Moroz, M.; Holland, A.M.; Yim, N.L.; Rao, U.K.; Young, L.F.; Tannenbaum, D.; Masih, D.; Velardi, E.; et al. Adoptively transferred TRAIL+ T cells suppress GVHD and augment antitumor activity. J. Clin. Investig. 2013, 123, 2654–2662. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Haas, T.L. Biochemical analysis of the native TRAIL death-inducing signaling complex. Methods Mol. Biol. 2008, 414, 221–239. [Google Scholar] [PubMed]
- Ashkenazi, A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat. Rev. Drug Discov. 2008, 7, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Budd, R.C.; Yeh, W.C.; Tschopp, J. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol. 2006, 6, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.E.; Krammer, P.H. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003, 10, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, J.; Bellail, A.; Jiang, W.; Hugh, J.; Kneteman, N.M.; Hao, C. Inhibition of RIP and c-FLIP enhances TRAIL-induced apoptosis in pancreatic cancer cells. Cell. Signal. 2007, 19, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, X.; Yu, H.; Wei, S.; Williams, N.; Holmes, D.L.; Halfmann, R.; Naidoo, J.; Wang, L.; Li, L.; et al. Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nat. Chem. Biol. 2013, 9, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kim, S.H.; El-Deiry, W.S. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc. Natl. Acad. Sci. USA 2006, 103, 11003–11008. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, E.; Sarhan, D.; Carlsten, M.; Kaminskyy, V.O.; D’Arcy, P.; Zhivotovsky, B.; Childs, R.; Lundqvist, A. Doxorubicin sensitizes human tumor cells to NK cell- and T-cell-mediated killing by augmented TRAIL receptor signaling. Int. J. Cancer 2013, 133, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Vitovski, S.; Chantry, A.D.; Lawson, M.A.; Croucher, P.I. Targeting tumour-initiating cells with TRAIL based combination therapy ensures complete and lasting eradication of multiple myeloma tumours in vivo. PLoS One 2012, 7, e35830. [Google Scholar] [CrossRef] [PubMed]
- Borbone, E.; Berlingieri, M.T.; de Bellis, F.; Nebbioso, A.; Chiappetta, G.; Mai, A.; Altucci, L.; Fusco, A. Histone deacetylase inhibitors induce thyroid cancer-specific apoptosis through proteasome-dependent inhibition of TRAIL degradation. Oncogene 2010, 29, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, M.L.; Idowu, M.O.; Zhao, Y.; Khalak, H.; Payne, K.K.; Wang, X.Y.; Dumur, C.I.; Bedognetti, D.; Tomei, S.; Ascierto, P.A.; et al. Molecular signatures mostly associated with NK cells are predictive of relapse free survival in breast cancer patients. J. Transl. Med. 2013, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.; Lundqvist, A.; McCoy, P., Jr.; Samsel, L.; Fan, Y.; Tawab, A.; Childs, R. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy 2009, 11, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Wennerberg, E.; Lundqvist, A. Unpublished work. 2014.
- Sarhan, D.; D’Arcy, P.; Wennerberg, E.; Liden, M.; Hu, J.; Winqvist, O.; Rolny, C.; Lundqvist, A. Activated monocytes augment TRAIL-mediated cytotoxicity by human NK cells through release of IFN-γ. Eur. J. Immunol. 2013, 43, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Xu, R.; Peach, M.; Huang, C.P.; Branstetter, D.; Novotny, W.; Herbst, R.S.; Eckhardt, S.G.; Holland, P.M. Evaluation of pharmacodynamic biomarkers in a Phase 1a trial of dulanermin (rhApo2L/TRAIL) in patients with advanced tumours. Br. J. Cancer 2011, 105, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Vose, J.M.; Zelenetz, A.D.; Smith, M.R.; Burris, H.A.; Ansell, S.M.; Klein, J.; Halpern, W.; Miceli, R.; Kumm, E.; et al. A Phase 1b/2 trial of mapatumumab in patients with relapsed/refractory non-Hodgkin’s lymphoma. Br. J. Cancer 2010, 103, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.S.; Geller, J.I.; Baird, K.; Chou, A.J.; Galli, S.; Charles, A.; Amaoko, M.; Rhee, E.H.; Price, A.; Wexler, L.H.; et al. Phase I trial and pharmacokinetic study of lexatumumab in pediatric patients with solid tumors. J. Clin. Oncol. 2012, 30, 4141–4147. [Google Scholar] [PubMed]
- Zhang, X.D.; Nguyen, T.; Thomas, W.D.; Sanders, J.E.; Hersey, P. Mechanisms of resistance of normal cells to TRAIL induced apoptosis vary between different cell types. FEBS Lett. 2000, 482, 193–199. [Google Scholar] [PubMed]
- Zhang, Y.; Zhang, B. TRAIL resistance of breast cancer cells is associated with constitutive endocytosis of death receptors 4 and 5. Mol. Cancer Res. 2008, 6, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Ebinger, M.; Senf, L.; Wachowski, O.; Scheurlen, W. Promoter methylation pattern of caspase-8, P16INK4A, MGMT, TIMP-3, and E-cadherin in medulloblastoma. Pathol. Oncol. Res. 2004, 10, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Hopkins-Donaldson, S.; Ziegler, A.; Kurtz, S.; Bigosch, C.; Kandioler, D.; Ludwig, C.; Zangemeister-Wittke, U.; Stahel, R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell. Death Differ. 2003, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Anderson, K.C.; Treon, S.P. Intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human multiple myeloma cells. Blood 2002, 99, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Zang, F.; Wei, X.; Leng, X.; Yu, M.; Sun, B. C-FLIP(L) contributes to TRAIL resistance in HER2-positive breast cancer. Biochem. Biophys. Res. Commun. 2014, 450, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ili, C.G.; Brebi, P.; Tapia, O.; Sandoval, A.; Lopez, J.; Garcia, P.; Leal, P.; Sidransky, D.; Guerrero-Preston, R.; Roa, J.C. Cellular FLICE-like inhibitory protein long form (c-FLIPL) overexpression is related to cervical cancer progression. Int. J. Gynecol. Pathol. 2013, 32, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.R.; Min, K.J.; Kim, S.; Park, J.W.; Park, W.K.; Lee, T.J.; Kwon, T.K. Anisomycin treatment enhances TRAIL-mediated apoptosis in renal carcinoma cells through the down-regulation of Bcl-2, c-FLIP(L) and Mcl-1. Biochimie 2013, 95, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Penington, R.C.; Tang, X.M. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis is inhibited by Bcl-2 but restored by the small molecule Bcl-2 inhibitor, HA 14–1, in human colon cancer cells. Clin. Cancer Res. 2004, 10, 8284–8292. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Trauzold, A.; Boenicke, L.; Sandberg, C.; Beckmann, S.; Bayer, E.; Walczak, H.; Kalthoff, H.; Ungefroren, H. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene 2000, 19, 5477–5486. [Google Scholar] [CrossRef] [PubMed]
- Kulathu, Y.; Komander, D. Atypical ubiquitylation—The unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Song, J.J.; Szczepanski, M.J.; Kim, S.Y.; Kim, J.H.; An, J.Y.; Kwon, Y.T.; Alcala, M.A., Jr.; Bartlett, D.L.; Lee, Y.J. c-Cbl-mediated degradation of TRAIL receptors is responsible for the development of the early phase of TRAIL resistance. Cell. Signal. 2010, 22, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Van de Kooij, B.; Verbrugge, I.; de Vries, E.; Gijsen, M.; Montserrat, V.; Maas, C.; Neefjes, J.; Borst, J. Ubiquitination by the membrane-associated RING-CH-8 (MARCH-8) ligase controls steady-state cell surface expression of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) receptor 1. J. Biol. Chem. 2013, 288, 6617–6628. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Li, Y.; Pitti, R.; Lawrence, D.; Pham, V.C.; Lill, J.R.; Ashkenazi, A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 2009, 137, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Bellail, A.C.; Olson, J.J.; Yang, X.; Chen, Z.J.; Hao, C. A20 ubiquitin ligase-mediated polyubiquitination of RIP1 inhibits caspase-8 cleavage and TRAIL-induced apoptosis in glioblastoma. Cancer Discov. 2012, 2, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kong, Y.; Zhou, Z.; Chen, H.; Wang, Z.; Hsieh, Y.C.; Zhao, D.; Zhi, X.; Huang, J.; Zhang, J.; et al. The HECTD3 E3 ubiquitin ligase facilitates cancer cell survival by promoting K63-linked polyubiquitination of caspase-8. Cell Death Dis. 2013, 4, e935. [Google Scholar] [CrossRef] [PubMed]
- Christian, P.A.; Fiandalo, M.V.; Schwarze, S.R. Possible role of death receptor-mediated apoptosis by the E3 ubiquitin ligases Siah2 and POSH. Mol. Cancer 2011, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, R.; Chen, C. The WWP1 ubiquitin E3 ligase increases TRAIL resistance in breast cancer. Int. J. Cancer 2012, 130, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIP(L) turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef] [PubMed]
- De Wilt, L.H.; Kroon, J.; Jansen, G.; de Jong, S.; Peters, G.J.; Kruyt, F.A. Bortezomib and TRAIL: A perfect match for apoptotic elimination of tumour cells? Crit. Rev. Oncol./Hematol. 2013, 85, 363–372. [Google Scholar] [CrossRef]
- Liu, X.; Yue, P.; Chen, S.; Hu, L.; Lonial, S.; Khuri, F.R.; Sun, S.Y. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res. 2007, 67, 4981–4988. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, K.; Kraft, A.S. Proteasome inhibitor PS-341 (VELCADE) induces stabilization of the TRAIL receptor DR5 mRNA through the 3'-untranslated region. Mol. Cancer Ther. 2008, 7, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Chou, C.W.; Chang, Y.F.; Chen, C.C. Proteasome inhibitors enhance TRAIL-induced apoptosis through the intronic regulation of DR5: Involvement of NF-κB and reactive oxygen species-mediated p53 activation. J. Immunol. 2008, 180, 8030–8039. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Seol, J.W.; Jeong, J.K.; Moon, M.H.; Park, S.Y. Inhibition of the ubiquitin-proteasome system sensitizes TRAIL-resistant prostate cancer cells by up-regulation of death receptor 5. Mol. Med. Rep. 2011, 4, 1255–1259. [Google Scholar] [PubMed]
- Johnson, T.R.; Stone, K.; Nikrad, M.; Yeh, T.; Zong, W.X.; Thompson, C.B.; Nesterov, A.; Kraft, A.S. The proteasome inhibitor PS-341 overcomes TRAIL resistance in Bax and caspase 9-negative or Bcl-xL overexpressing cells. Oncogene 2003, 22, 4953–4963. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Bradner, J.E.; Wong, J.; Chauhan, D.; Richardson, P.; Schreiber, S.L.; Anderson, K.C. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. USA 2005, 102, 8567–8572. [Google Scholar] [CrossRef] [PubMed]
- Unterkircher, T.; Cristofanon, S.; Vellanki, S.H.; Nonnenmacher, L.; Karpel-Massler, G.; Wirtz, C.R.; Debatin, K.M.; Fulda, S. Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBID stability and mitochondrial apoptosis. Clin. Cancer Res. 2011, 17, 4019–4030. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, A.; Abrams, S.I.; Schrump, D.S.; Alvarez, G.; Suffredini, D.; Berg, M.; Childs, R. Bortezomib and depsipeptide sensitize tumors to tumor necrosis factor-related apoptosis-inducing ligand: A novel method to potentiate natural killer cell tumor cytotoxicity. Cancer Res. 2006, 66, 7317–7325. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, A.; Berg, M.; Smith, A.; Childs, R.W. Bortezomib treatment to potentiate the anti-tumor immunity of ex-vivo expanded adoptively infused autologous natural killer cells. J. Cancer 2011, 2, 383–385. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknas, M.; Lindsten, K.; de Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, P.; Linder, S. Proteasome deubiquitinases as novel targets for cancer therapy. Int. J. Biochem. Cell. Biol. 2012, 44, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yue, P.; Lonial, S.; Khuri, F.R.; Sun, S.Y. The NEDD8-activating enzyme inhibitor, MLN4924, cooperates with TRAIL to augment apoptosis through facilitating c-FLIP degradation in head and neck cancer cells. Mol. Cancer Ther. 2011, 10, 2415–2425. [Google Scholar] [CrossRef] [PubMed]
- Grund, K.; Ahmadi, R.; Jung, F.; Funke, V.; Gdynia, G.; Benner, A.; Sykora, J.; Walczak, H.; Joos, S.; Felsberg, J.; et al. Troglitazone-mediated sensitization to TRAIL-induced apoptosis is regulated by proteasome-dependent degradation of FLIP and ERK1/2-dependent phosphorylation of BAD. Cancer Biol. Ther. 2008, 7, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, A.; Su, S.; Rao, S.; Childs, R. Cutting edge: Bortezomib-treated tumors sensitized to NK cell apoptosis paradoxically acquire resistance to antigen-specific T cells. J. Immunol. 2010, 184, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarhan, D.; D'Arcy, P.; Lundqvist, A. Regulation of TRAIL-Receptor Expression by the Ubiquitin-Proteasome System. Int. J. Mol. Sci. 2014, 15, 18557-18573. https://doi.org/10.3390/ijms151018557
Sarhan D, D'Arcy P, Lundqvist A. Regulation of TRAIL-Receptor Expression by the Ubiquitin-Proteasome System. International Journal of Molecular Sciences. 2014; 15(10):18557-18573. https://doi.org/10.3390/ijms151018557
Chicago/Turabian StyleSarhan, Dhifaf, Padraig D'Arcy, and Andreas Lundqvist. 2014. "Regulation of TRAIL-Receptor Expression by the Ubiquitin-Proteasome System" International Journal of Molecular Sciences 15, no. 10: 18557-18573. https://doi.org/10.3390/ijms151018557