1. Introduction
Since its introduction in 1996, the use of commercial genetically modified (GM) crops has drastically expanded. In the year 2013, 175.2 million hectares of GM crops were grown. Of these, less than 0.3 million hectares were cultivated in the EU (Spain, Portugal, Czech Republic, Romania, and Slovakia) to one singular maize variety (MON810) [
1]. More recently, the commercialization of single events (for example MON810 maize or Roundup Ready Soy), have given way to the introduction of stacked events containing multiple transgenes [
2]. Approximately 47 million hectares of stacked traits were grown in 2013, which is 3.3 million hectares more than that reported in 2012 [
1]. There has been an increase in the number of approved stacked events and the number of transgenes stacked in a single plant (e.g., SmartStax™ maize, containing eight events) [
2].
Authorization and compliance measures of GM food and feed require that GM events be genetically characterized and quantified. Additionally, transgene stability is important aspect to ensure food safety [
3,
4]. Reports on the genetic stability of genetically modified organisms (GMOs) have presented variable results. Initially, it was presumed that the production of transgenic plants involved the insertion of a single transgene and the flanking regions without further genomic disruption. However, instances of genomic alterations, including complex nucleotide rearrangements (e.g., deletions, duplications, insertions of organelle and filler DNA, and translocations) have occurred as a result of the transformation process [
5,
6,
7,
8,
9,
10,
11,
12,
13]. The stability of the transgene, and gene expression as well as performance of the plant can be influenced by various factors, including host biology, genome organization, by epigenetic factors, or by nucleotide changes within the introduced DNA construct [
4].
In addition to the requirement of genetic stability during GMO development, transgenic plants should demonstrate genetic stability subsequently during cultivation and propagation. Recent studies have identified the occurrence of DNA rearrangements after the transformation process and during the post-release monitoring. Choffnes
et al. [
14] studied transgene integration patterns of several hundred plants in subsequent generations. It was demonstrated that the original transgene integration patterns of regenerated primary transgenic plants (T
0) were not stable when passed to progenies, possibly due to a high level of homologous recombination. Another study performed cytogenetic analyses of GM maize and revealed a fragile phenotype of 45S rDNA as a consequence of genetic modification [
15]. McCabe
et al. [
11] detected differences in transgene expression between T
0 plants and plants grown from selfed T
0 seeds (T
1 generation) of transgenic lettuce. And it was suggested that these differences were due to inactivation or suppression of transgene expression. Ulian
et al. [
16] reported instabilities of transfer DNA (T-DNA) insertions in T
0 plant genomes of petunia. Differences in plant protein expression that originated from different locations were also discovered [
17,
18].
In recent years, new methods have been adapted for profiling the epigenome, proteome, and transcriptome of GMOs [
19]. La Paz,
et al. [
20] analyzed cytosine methylation of the MON810 transgene in different MON810 varieties. The methylation level of the transgene was very low and a comparison between the different varieties revealed no significant differences in symmetric DNA methylation. In contrast, significant differences were observed in the asymmetric sites that play a minor role in epigenetics. Most of the reported comparative transcriptome analyses of MON810 maize were performed using microarrays [
21,
22,
23]. La Paz
et al. [
24] analyzed MON810 and isogenic maize varieties using high-throughput RNA sequencing and found 140 differentially expressed genes. The authors suggested that the differences were due to a slightly delayed maturation process of MON810 compared to the conventional varieties. A proteomic approach was performed by Agapito-Tenfen
et al. [
25]. Using two-dimensional gel electrophoresis combined with mass spectrometry, 32 differentially expressed proteins were identified in Brazilian MON810 maize when compared to isogenic control maize. Rang
et al. [
26] detected different RNA variants transcribed from the transgene in Roundup Ready soybean which may code for 5-Enol-pyruvylshikimate-3-phosphate synthase (EPSPS) fusion proteins.
According to the European Food Safety Authority (EFSA) guidance document and the EU directive 2001/18/EC, the introduced DNA construct must be stable and no changes in the DNA construct should occur during the cultivation and propagation of the plants [
27,
28]. Even minor changes in the construct are critical because they may lead to unintended changes in the plant properties, content, and/or morphology [
29]. Additionally, genetic heterogeneity in the samples may render the analysis for GMO detection unreliable or equivocal [
30]. The EFSA guidance document details the requirements for the authorization of GMOs. Accordingly, the GMO events must be analyzed for DNA rearrangements and instabilities of the insert [
27,
28]. Genetic stability is commonly verified by Southern Blot analysis, however, Southern Blots have drawbacks, including low sensitivity (e.g., only major DNA changes can be detected). In many cases minor changes may occur, which can have an impact on the plant. A variety of methods have been used for analyzing minor nucleotide changes. Genomic samples can be sequenced directly or GMOs can be analyzed by methods such as Sensitive Capillary Electrophoresis (CSCE) or long-range polymerase chain reaction (PCR). Further, real-time PCR employing Scorpion primers for amplification or high resolution melting (HRM) analysis can be used to distinguish between different alleles or to filter out mutated samples [
31,
32].
According to EFSA, applicants must demonstrate the genetic stability of the transgenic locus over five generations or vegetative cycles before a product can be authorized. For stacked events, the applicant must establish the integrity of the inserts [
33]. Further, EFSA determined that the risk assessment of stacked events consisting of events approved by the EU should focus on the genetic stability of such plants, the expression level of the transgenes, and unintended interactions between the stacked events [
34]. The genetic stability of the single insert is verified by Southern Blot and segregation analyses before authorization [
4,
35,
36]. The EU directive 2001/18/EC requires that the GMOs be inspected every 10 years [
28]. Aguilera
et al. recommended the validation of the genetic stability of inserts for the whole lifespan of a product [
37].
In this study, the genetic stability of a stacked maize (MON88017 × MON810) and that of a single oilseed rape event (GT73) were investigated. MON88017 × MON810 is a maize variety produced by conventional crossing of a double-stacked maize (MON88017) that expresses a
CP4 EPSPS gene from Agrobacterium sp. CP4, conferring resistance to glyphosate, and the coleopteran-active delta-endotoxin Cry3Bb1 gene from Bacillus thuringiensis (Bt) (
cry3Bb1) to provide protection against coleopteran insects, particularly the maize rootworm, and the lepidopteran-active delta-endotoxin Cry1Ab (
cry1Ab)-containing MON810 for protection against lepidopterans such as
Ostrinia nubilalis and
Sesamia spp. GT73 is a glyphosate-tolerant oilseed rape [
38]. We used real-time PCR with Scorpion primers for amplification (Scorpion PCR) and HRM analysis to examine whether DNA alterations occurred amongst individual seeds (of MON88017 × MON810 maize) or plants (of GT73 oilseed rape). Earlier studies have adapted these methods to analyze the genetic stability and organization of transgenic insertion events. In Neumann
et al. [
32] it could be shown that single nucleotide polymorphism (SNPs) in plasmids can be demonstrated by Scorpion analysis. As a further work for optimizing the method, different alleles of the alcohol dehydrogenase 1 (
ADH1) gene in maize were investigated by Madi
et al. [
31]. In single seeds, the homozygous as well as heterozygous state of the
ADH1 gene was found. Alleles can be discriminated using Scorpion PCR as well as by HRM analysis. They can be separated by cloning the PCR-amplified DNA. Testing of these clones allows a better differentiation between the alleles compared to testing only the genomic DNA.
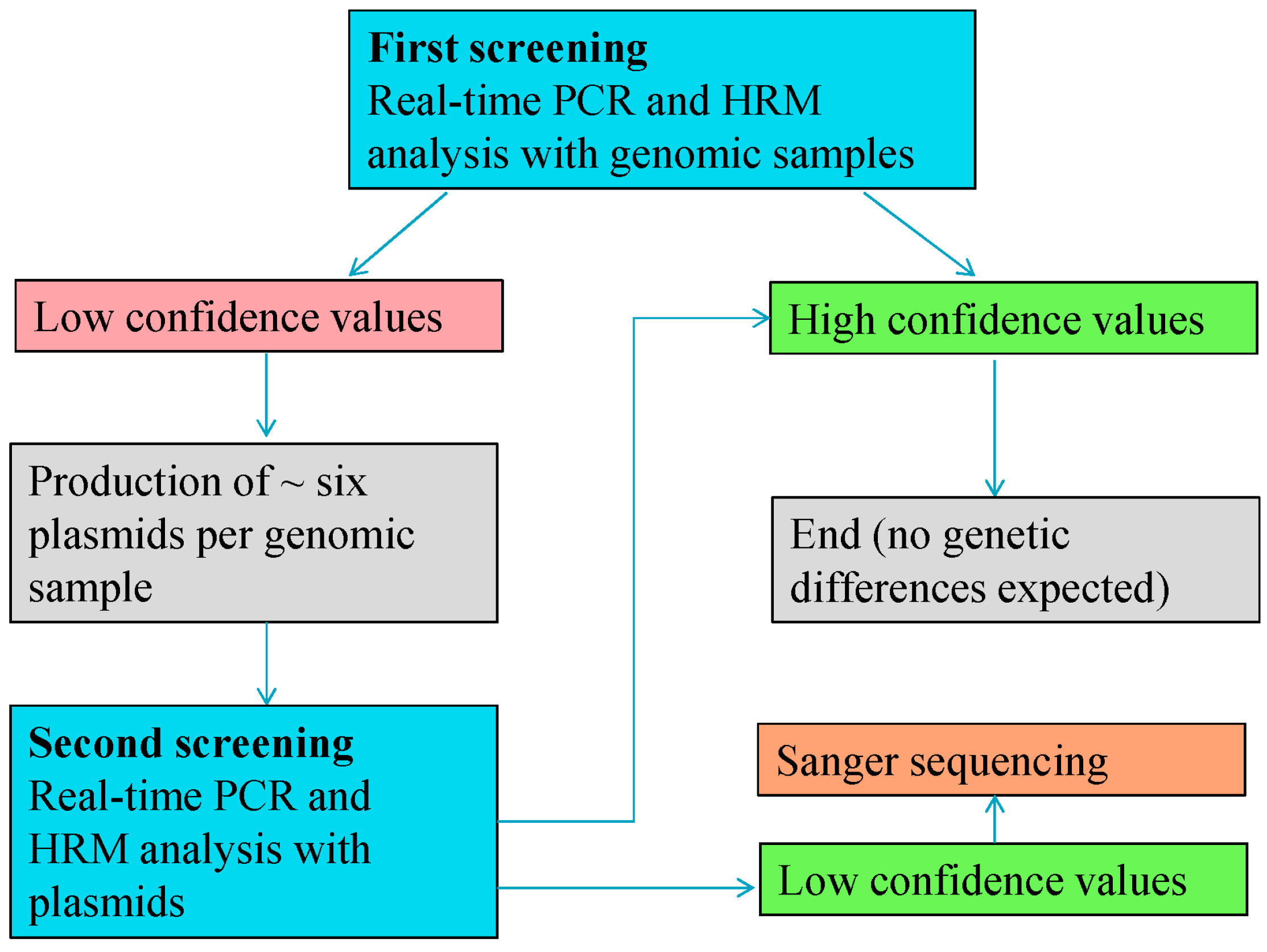
In the present work, real-time PCR in combination with subsequent HRM analysis is used as a highly sensitive mutation scanning of selected DNA regions as a standardized means for determining the level of the genetic stability. Samples that potentially carried mutations were cloned into plasmids and sequenced by the Sanger method to verify the DNA alterations.
3. Experimental Section
Unless stated otherwise, the chemicals, PCR reagents, and PCR primers used were of analytical grade and were purchased from Biozym (Hessisch Oldendorf, Germany) (PCR reagents) and Sigma–Aldrich (Vienna, Austria) (PCR primers and chemicals). The Zero Blunt TOPO PCR Cloning kit (Invitrogen, Vienna, Austria), EasyPrep Pro Plasmid Miniprep kit (Biozym), JM109 Competent Cells (>107 cfu/μg, Promega, Mannheim, Germany), Wizard DNA Clean-up System (Promega), Phusion Hot Start II High-Fidelity DNA Polymerase kit (Biozym), and Type-it HRM PCR Kit (Qiagen, Hilden, Germany) were used according to the respective manufacturer’s recommendations. The stacked event MON88017-3 × MON-00810-6 (hybrid 4421VT3) maize was obtained from Croplan Genetics® (Williston, ND, USA). And the GT73 (MON-00073-7) oilseed rape seeds were obtained by taking plants randomly from a field in Saskatchewan, Canada (season 2011). GT73 oilseed rape plants were cultivated in a closed system (in a separate room under controlled conditions: day/night 20 ± 2 °C, 12/12 h light/dark, r. h. 60% ± 5%). Leaves were harvested after four weeks and immediately frozen in liquid nitrogen.
3.2. Instruments Used for HRM and Scorpion Analysis
Scorpion PCR experiments can be performed on any standard real-time PCR instrument. For HRM analysis, it is necessary to use a specific cycler optimized for precise melting analysis that uses dedicated heating algorithms and specific software [
45,
46]. For the Scorpion PCR as well as for the HRM experiments a Rotor-Gene Q (Qiagen) instrument was used. For the calculation of the scores (confidence values) of the HRM analysis, a specific software package (Rotor-Gene 2.0.2.4, Qiagen) of the Rotor-Gene Q cycler was used.
3.3. Screening by Real-Time PCR Using Scorpion Primers and HRM Analysis
A total 510 individual oilseed rape samples (GT73) and 100 individual maize samples (MON88017 × MON810) were analyzed on different days by three different operators. The mean of the values was calculated and was used for further investigation. Pools of genomic samples (for primary screenings) or plasmids (secondary screenings) served as reference to calculate the confidence values. The reference served as a control for the performance of all samples (including a positive (standard) control as well as a non-template control), allowed for the verification of the validity of all parameters of the run, and provided a reference value that enabled a comparison of different runs.
Unimolecular, singular Scorpion primers consisting of forward/reverse PCR-primer sequences as well as additional elements were used for this work. The additional elements included a PCR stopper to prevent the PCR read-through of the probe element, and a specific probe sequence with a fluorescence-based detection module consisting of a fluorophore and a quencher. The PCR yields an amplicon containing a sequence that is complementary to the Scorpion probe. The specific probe sequence then binds to its complementary sequence within the amplicon, the hairpin loop opens up, and a signal is produced [
32,
39].
With HRM analysis, the melting point (
Tm) of the amplicon produced is determined after the PCR reaction. This is achieved by gradually increasing the temperature from the annealing temperature up to 95 °C. Simultaneously, the fluorescence of a DNA intercalating dye is measured. Because high-precision temperature control and fluorescence measurement are required for successfully implementing this method, such measurements can only be made using an HRM-certified equipment [
40].
3.4. Qualitative PCR for Cloning
The PCR was performed with genomic DNA as template. A total 20 µL of the reaction mixture contained 4 µL of 5× Phusion HF-buffer (proprietary information, 1.5 mM MgCl
2, error rate was 4.4 × 10
−7), 500 nM of each primer, 250 µM of each deoxynucleoside triphosphate (dNTP), 0.4 U Phusion Hot Start II DNA Polymerase, and 50 ng of DNA. The temperature profile is shown in
Table 12.
Table 12.
The PCR temperature profile of the two-step protocol used for analysis. Touchdown PCR was used to eliminate non-specific amplification. Therefore, the annealing temperature started at 66 °C and was lowered by 0.5 °C per cycle for the first 18 cycles. The last 22 cycles had an annealing temperature of 60 °C.
Table 12.
The PCR temperature profile of the two-step protocol used for analysis. Touchdown PCR was used to eliminate non-specific amplification. Therefore, the annealing temperature started at 66 °C and was lowered by 0.5 °C per cycle for the first 18 cycles. The last 22 cycles had an annealing temperature of 60 °C.
| Cycle Step | Temp. | Time | Number of Cycles |
|---|
| Initial denaturation | 98 °C | 2 min | |
| Denaturation | 98 °C | 10 s | 18 |
| Annealing | 66–57.5 °C Touchdown (−0.5 °C each cycle) | 30 s |
| Extension | 72 °C | 30 s |
| Denaturation | 98 °C | 10 s | 22 |
| Annealing | 60 °C | 30 s |
| Extension | 72 °C | 30 s |
| Final extension | 72 °C | 7 min | |
A 1 µL aliquot of the PCR product was ligated and cloned into pCR-Blunt II-TOPO using the Zero Blunt TOPO PCR Cloning kit (Invitrogen) and transformed into JM109 Competent Cells. Colonies were grown in 5 mL of LB-medium and the recombinant DNA was isolated with the help of EasyPrep Pro Plasmid Miniprep kit (Biozym). Positive colonies were analyzed for the correct length of inserts using agarose gel electrophoresis on 2.5% gels. The validity of the insert was then verified by sequencing using M13 primers.
3.5. Real-Time PCR and HRM Conditions
Real-time PCR was performed on a Rotor-Gene Q (Qiagen) instrument. The total volume of the reaction mixture was 16 µL. The Type-it HRM PCR Kit (Qiagen), 700 nM primers (Scorpion primer 125 nM), and 1.6 µL of undiluted DNA (70–100 ng/µL) template were used for each reaction. The thermal cycling profile used is shown in
Table 13.
Table 13.
The thermal cycling profile of the real-time PCR (prior to HRM analysis) experiment.
Table 13.
The thermal cycling profile of the real-time PCR (prior to HRM analysis) experiment.
| Cycle Step | Temperature | Time | Number of Cycles |
|---|
| Initial Denaturation | 95 °C | 5 min | |
| Denaturation | 95 °C | 10 s | 55 |
| Annealing | 55 °C | 30 s |
| Extension | 72 °C | 20 s |
The experiments were performed on different days and the mean of the values was calculated and was used for further analysis. HRM was performed with the temperature increasing at a rate of 0.2 °C per 4 s. The initial and final temperatures were 70 and 95 °C, respectively. The melting curves and the temperature-shifted curves were normalized to enable the comparison of samples. Modified curves and HRM scores were obtained using the Rotor-Gene Q series software (version 2.0.2.4, Qiagen). The normalized and temperature-shifted melting curves corresponded to the final curve after normalization. When an amplicon harbored a sequence variation, the normalized and temperature-shifted melting curves had a different shape from those of the wild-type amplicons (see
Supplementary Figures S3 and S4).
3.6. Sequencing
All sequence analyses were performed using the Sanger sequencing method. The BigDye Fast cycle sequencing protocol, described by Platt
et al. [
47], was used for this purpose. The M13 sequences necessary for sequencing were present in the plasmids generated, as the cloning vector contained these sequences. To avoid sequencing errors, both forward and reverse sequencing were included in the method. This insured the reliability of the sequencing results.
3.7. Primers
Primers used for this work are shown
Table 1.
4. Conclusions
The objective of the present work was to identify DNA alterations that may cause genetic instability in GMOs, particularly in stacked events. The number of mutated samples we found in our study was two (out of 100 stacked maize samples tested). A much higher number of samples was analyzed (
n = 567 MON810 maize samples and
n = 1034 RRS 40-3-2 samples of soy) in earlier studies that found no DNA alterations in soybean and maize [
31,
32]. The SNP frequency we observed in our study markedly exceeds the natural mutation rate. The average single-base substitution rate for maize is estimated to be 5.39 × 10
−8 per site per year and for genic maize regions, 4.79 × 10
−8 per site per year [
48]. One important point to mention is that the detected SNPs were most likely not identified by low-sensitivity methods such as Southern Blot analysis.
Increased rate of mutation at the border regions may have a negative influence on GMO quantification by real-time PCR. Madi
et al. [
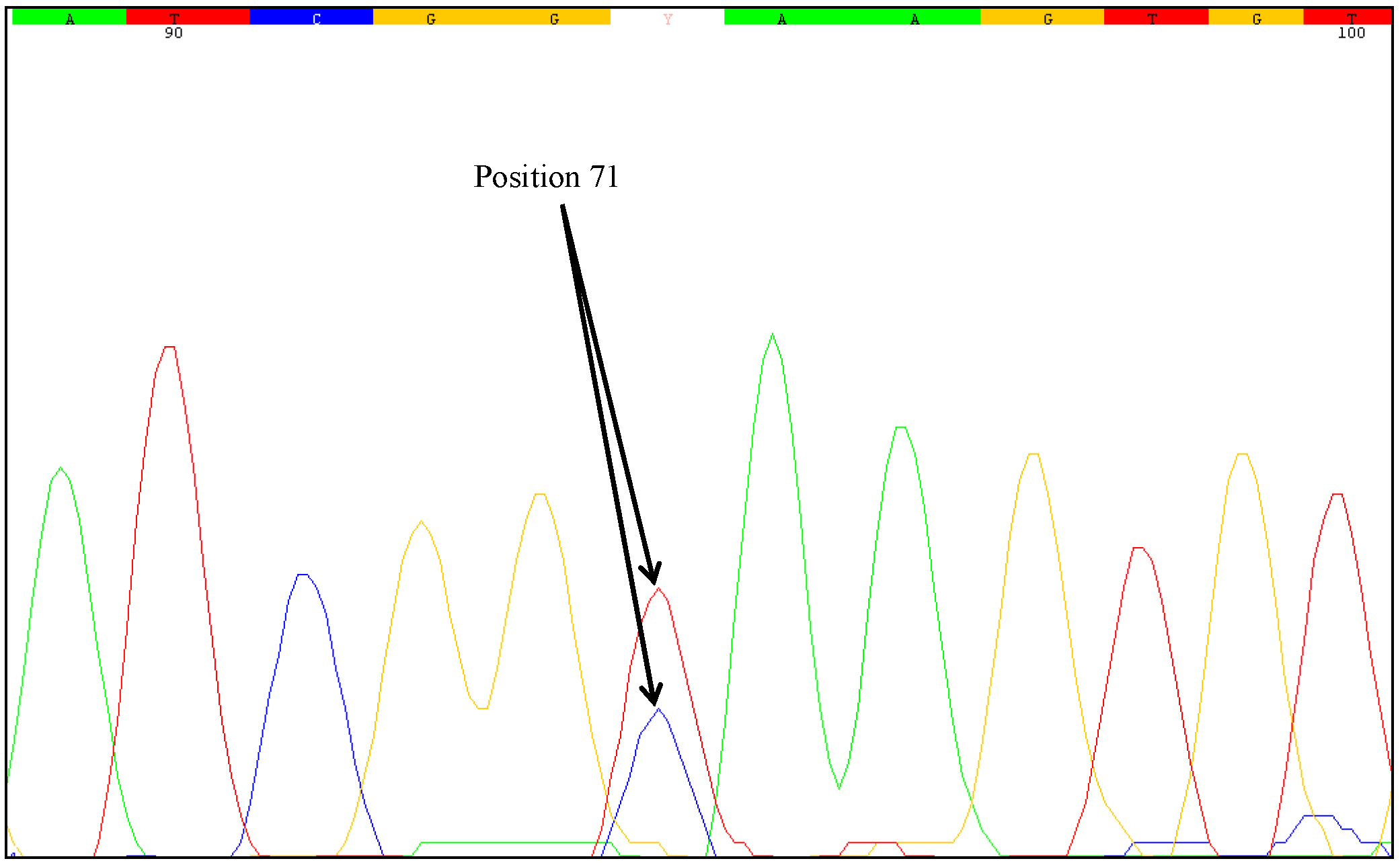
31] concluded that analysis of DNA instabilities in regions commonly used for the quantification of GMOs is particular important. Therefore, in canola and maize, the border regions from the genome into the construct at the 5' and 3' regions of the relevant transgene were tested in our study. Mutations were detected only in the 3'-MON810 region. Therefore, the 5'-MON810 region appears to be more suitable for GMO quantification as no instabilities were identified in this region. Based on the definition that the first base (at the 5' end) of the analyzed 3'-MON810 target region is at position one, we found that both of the mutated samples harbored an SNP at position 71. However, by investigating a higher number of stacked maize samples (MON88017 × MON810), it cannot be excluded that SNPs would be identified at (other) positions which may change the transgenic protein. The inserted 3' flanking region of MON810 is a truncated version of MON810. The 3' end of the
cry1Ab gene and the nopaline synthase (NOS) terminator were lost during the plant transformation [
49]. It is also possible that the occurrence of the SNP detected in this study at the same region was random. One additional aim of this study was to evaluate the impact of the identified SNP on the amino acid sequence. Similar to that reported by Ogasawara
et al. [
44], the identified mutation was silent, meaning the amino acid sequence remained unchanged. Therefore, it is conceivable that the phenotype of the sample was not altered.
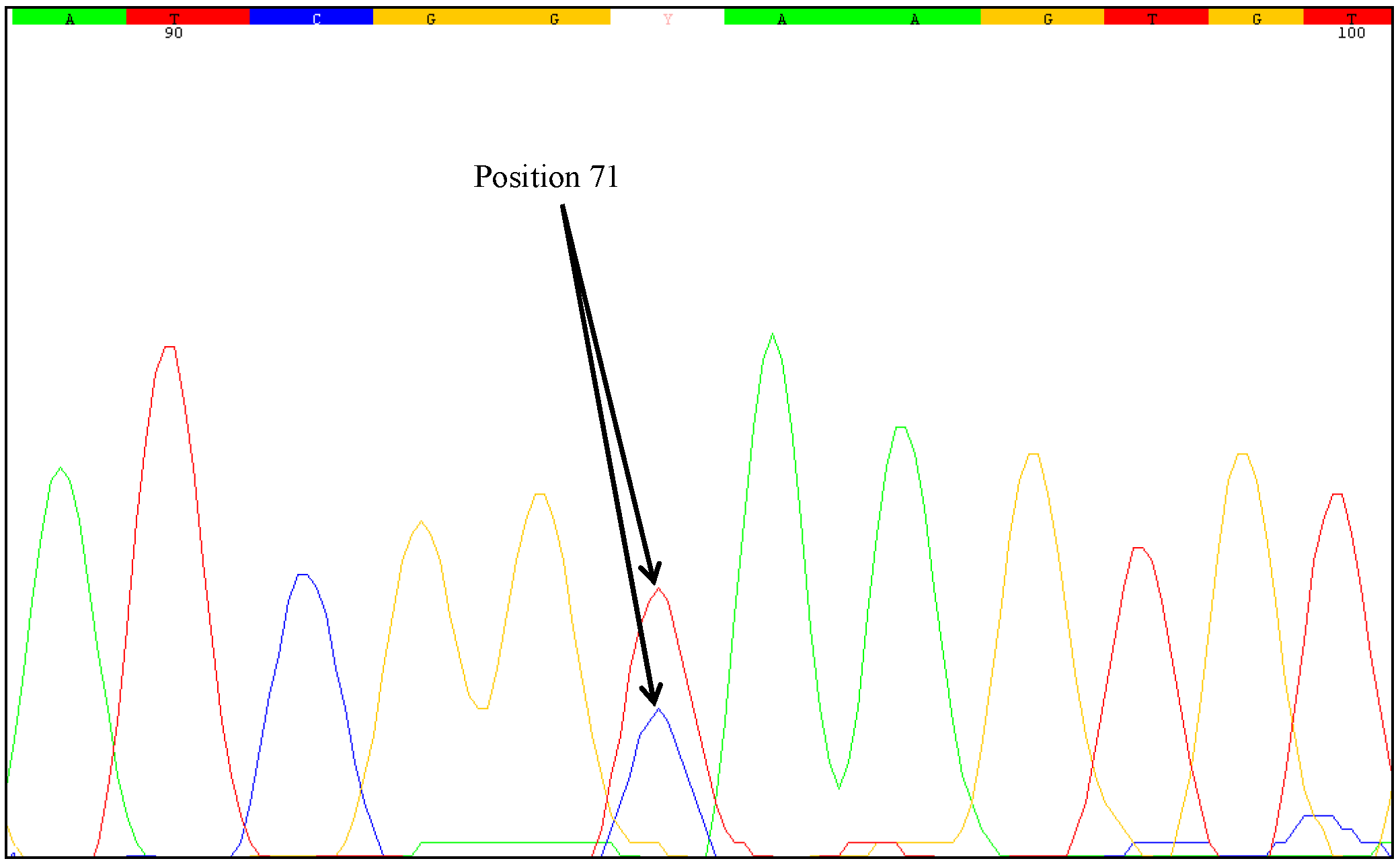
One unresolved question is the degree of zygosity of the transgene in the samples examined. Direct sequencing of the PCR products generated from the mutated samples indicated heterozygosity at nucleotide position 71. Two different nucleotides were identified at this position, a cytosine as wild-type and a thymine as mutant-type. The sequencing chromatograms of the PCR products clearly showed an overlay of these bases. However, sequencing errors could lead to similar chromatograms. Some positions may show overlapping nucleotides and it can be difficult to recognize heterozygous mutations just by direct sequencing of the PCR products. Therefore, we used the cloning approach described by Madi
et al. [
31]. Plasmids containing DNA of one allele can be obtained from genomic samples with heterozygous alleles. With clones comprised of one allele, the outcome of sequencing is unambiguous and sequencing error can be ruled out. PCR products of the affected samples were sequenced twice (forward and reverse). The plasmids of the MON88017 × MON810 samples 264 and 383 were also sequenced twice.
In Neumann
et al. [
32] and Madi
et al. [
31] the question was asked if the scores or the distribution of the values of the primary tests, (
i.e., the
Ct values of the Scorpion PCR and the confidence values of the HRM analyses) can be used to detect mutations. Our results show that the distribution of the values does not necessarily indicate the presence of mutations (
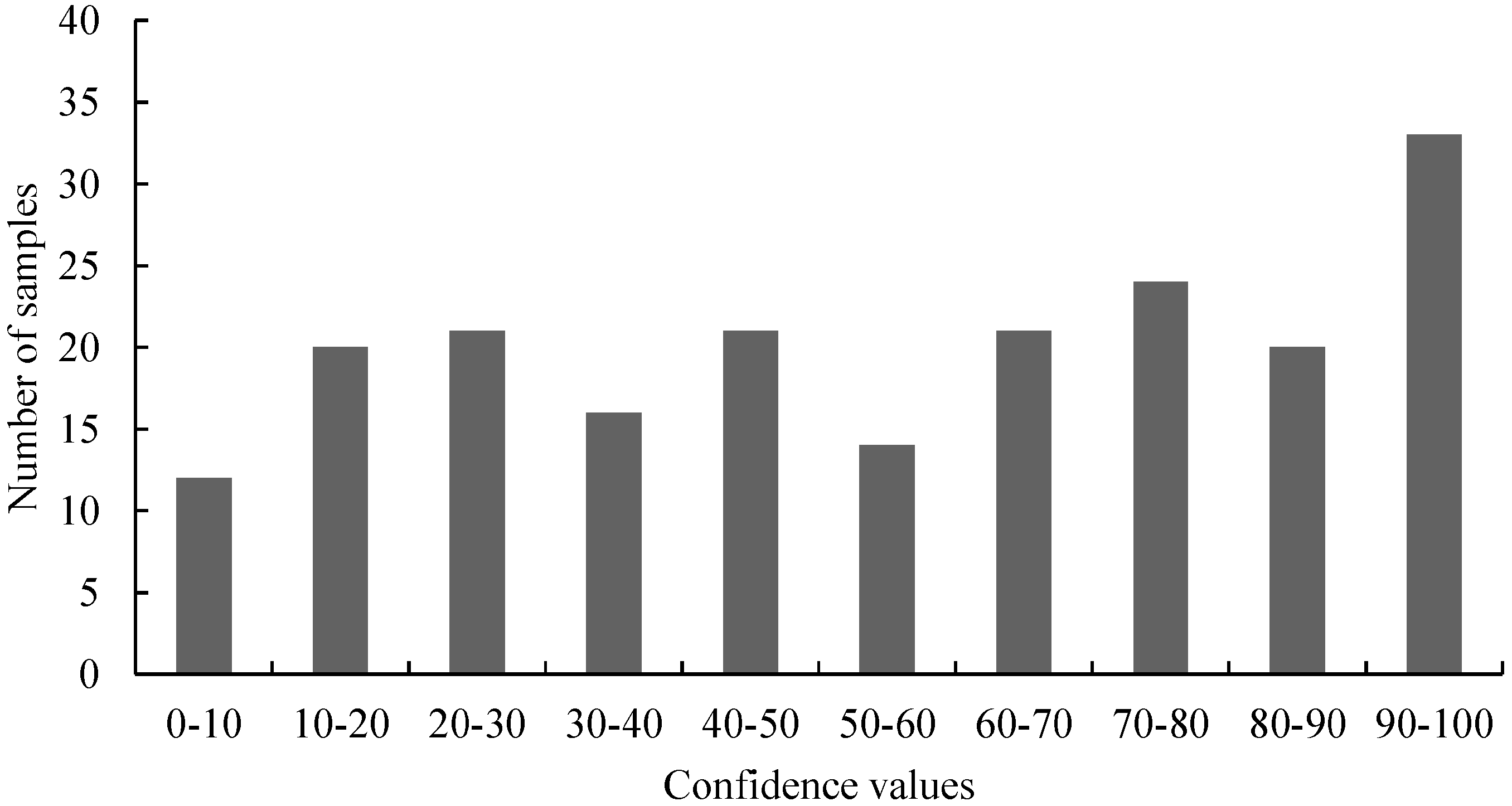
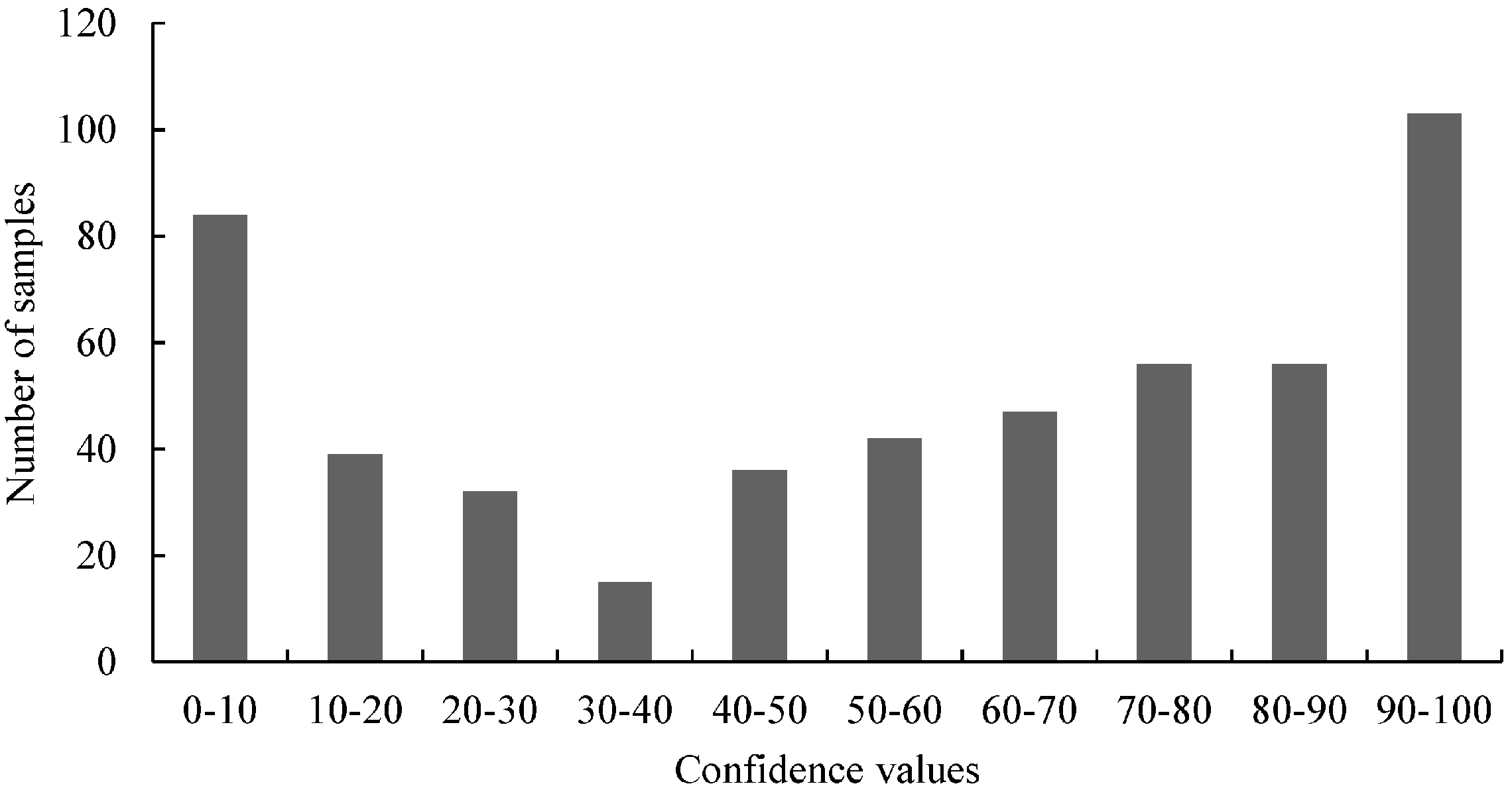
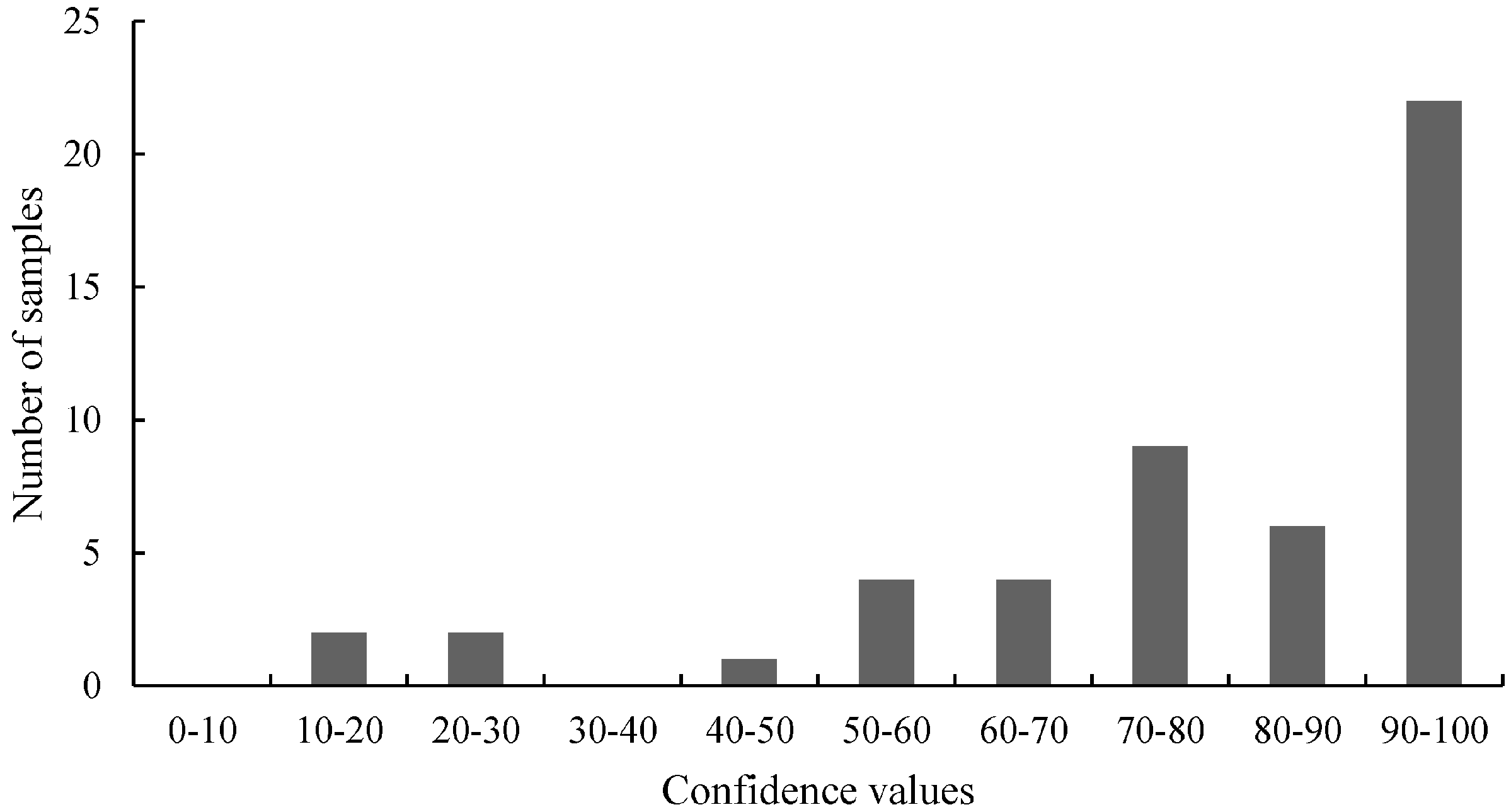
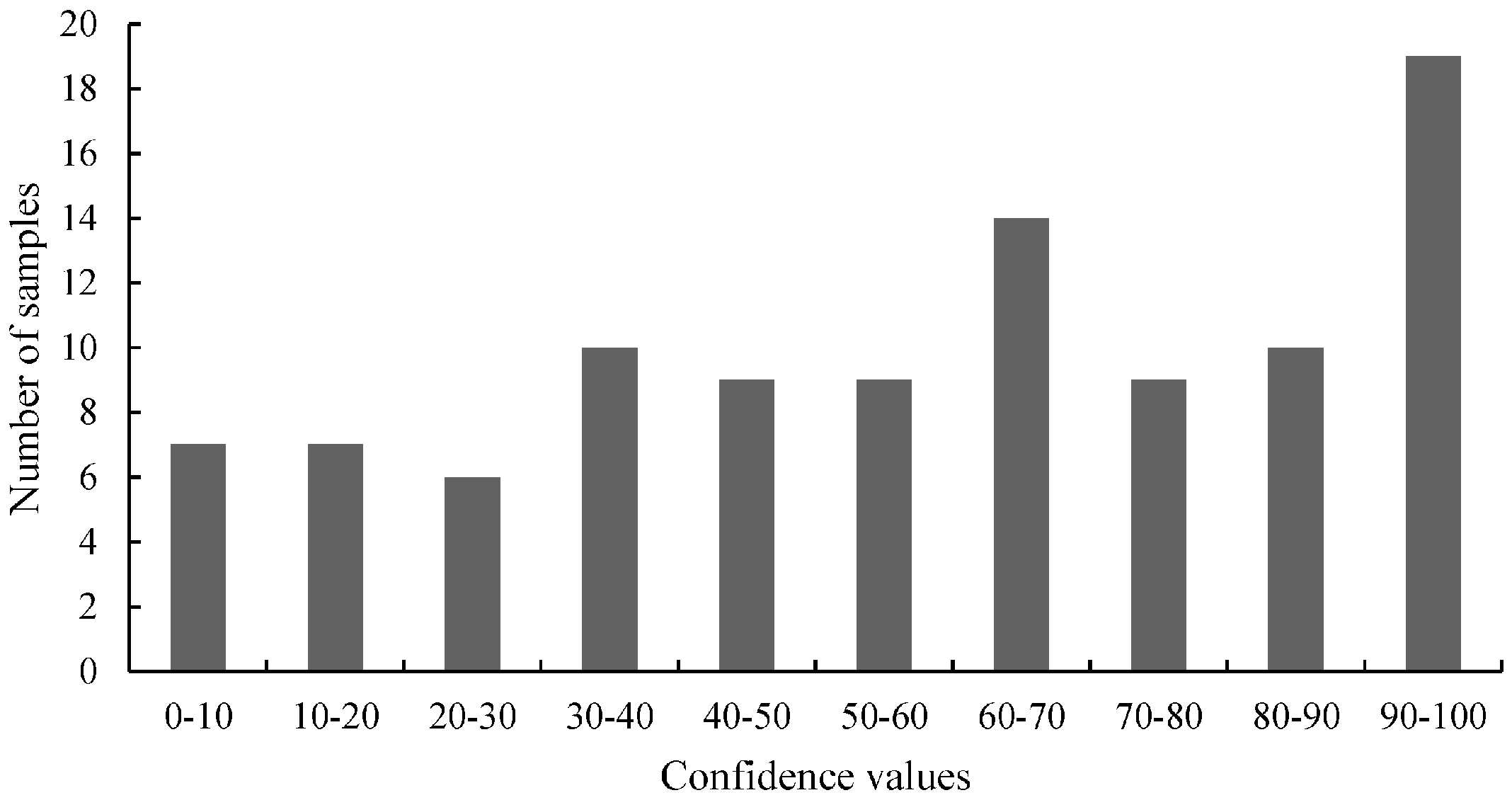
Figure 2,
Figure 4,
Figure 6 and
Figure 7). For samples harboring mutations, two groups can be expected: samples with very high confidence values (wild-type) and those with very low confidence values (mutated samples). However, only
Figure 4 (belongs to the distribution of 3'-GT73 samples) showed a distribution consistent with this description and no mutations were identified in these samples. In contrast, the distribution of the values of the 3'-MON810 screening did not show the expected characteristics. However, mutations were found in the 3'-MON810 region.
Based on the mutations identified by this work, we conclude that cloning is required for the unambiguous characterization of SNPs in GMOs. In addition to the two mutations, we also encountered false positive results revealed through subsequent investigation. The different plasmids of the affected sample (354 of MON88017 × MON810 maize) showed inconsistent SNPs (
Table 11). Although PCR error is a possible explanation, the precise nature of the factors that contributed to the false positive result is not known. Additionally, plasmid 275-8 of the 5'-MON810 region showed an SNP, which was assessed as due to error in the cloning sequence. A related aspect is the number of plasmids required. Our results show that minimum six to 10 plasmids are necessary for the characterization of heterozygous samples.
Whereas earlier studies investigated single MON810 maize events [
20,
32], the present work focused on stacked maize event containing MON810. Stacked events having the same promoter are more susceptible to unintended effects of the transgenic event expression [
50]. Many stacked events carry transgenes that use the same promoter. The 35S promoter, a viral element, occurs twice in MON88017 × MON810 maize. Kohli
et al. discussed that the 35S promoter contains a recombination hot spot that has an influence on GMOs [
51]. Since stacked events contain multiple viral promoters the susceptibility to instabilities may be increased. Due to an increasing number of stacked events being commercialized, there is a proportionally-relevant need for the analysis of their genetic stability in the context of the specific stacked event in question on a case-by-case basis. Due to the present results one might assume that stacked events tend to be more instable than single events. To confirm this assumption, it would be necessary to demonstrate further instabilities of MON810 in other stacked events. Corresponding experiments are planned. In these, it will be important to analyze whether stacking of the transgenes, and the means by which the stacked varieties were created, may be a contributing factor. It also will be necessary to understand the potential characterization (regulatory) and biosafety issues that may be relevant.
For future studies, it may also be interesting to analyze the zygosity degree of the mutated samples in detail.




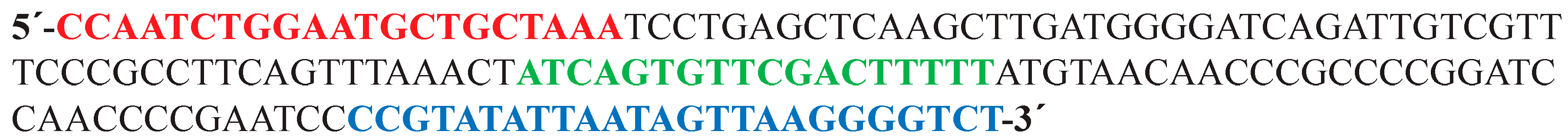{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}