The Effects of NAD+ on Apoptotic Neuronal Death and Mitochondrial Biogenesis and Function after Glutamate Excitotoxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
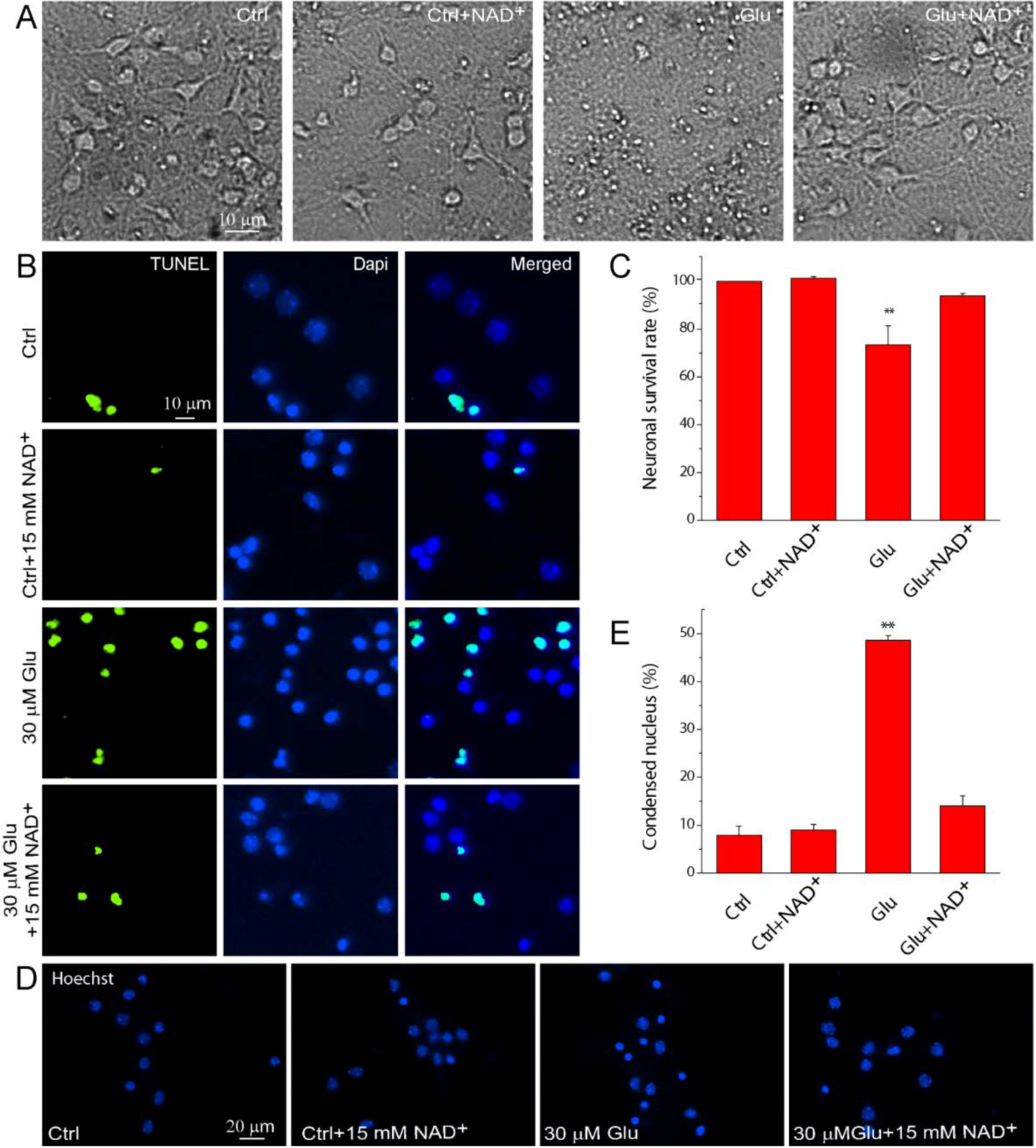
2.1. NAD+ Ameliorates Apoptotic Neuronal Death after Glutamate Stimulation
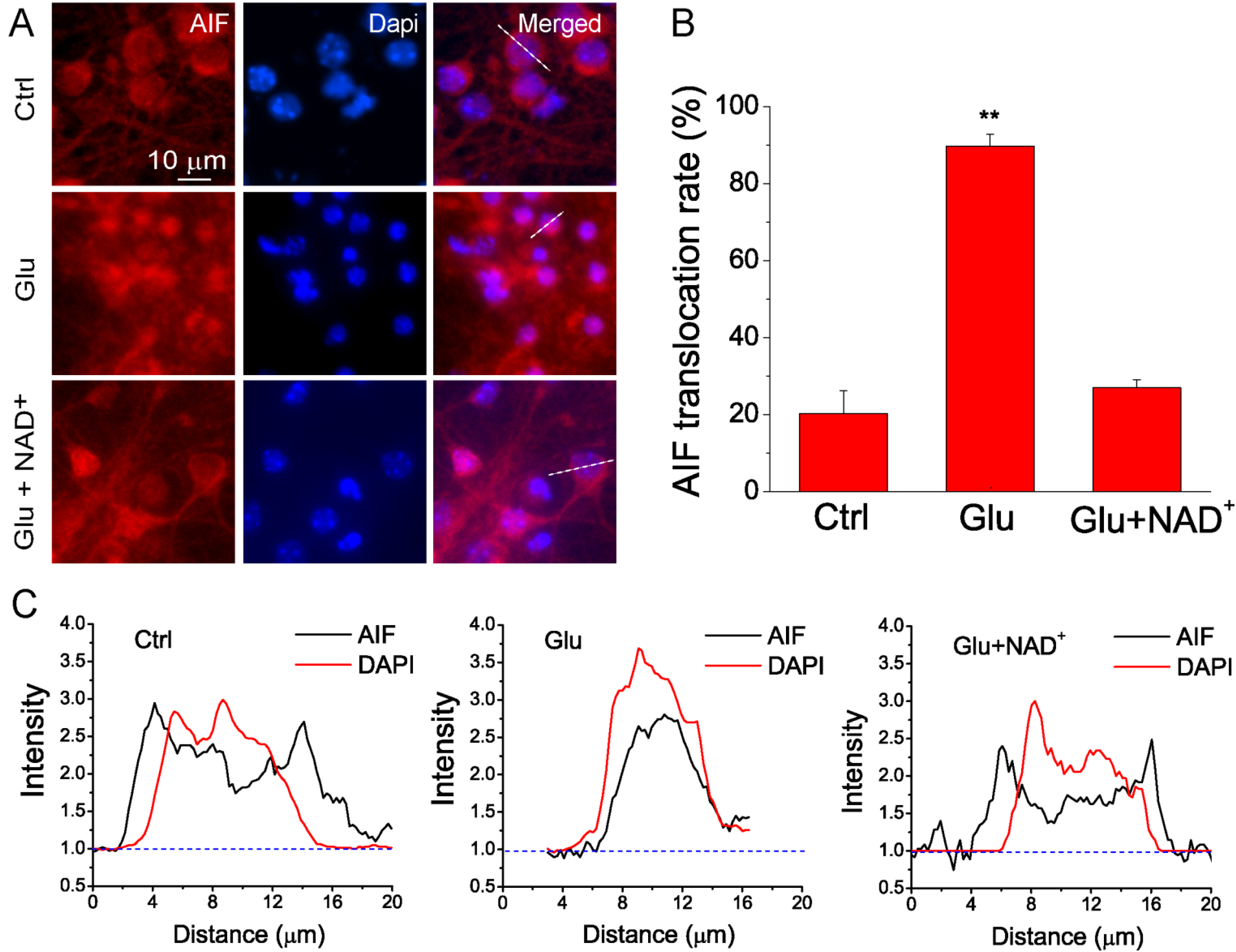
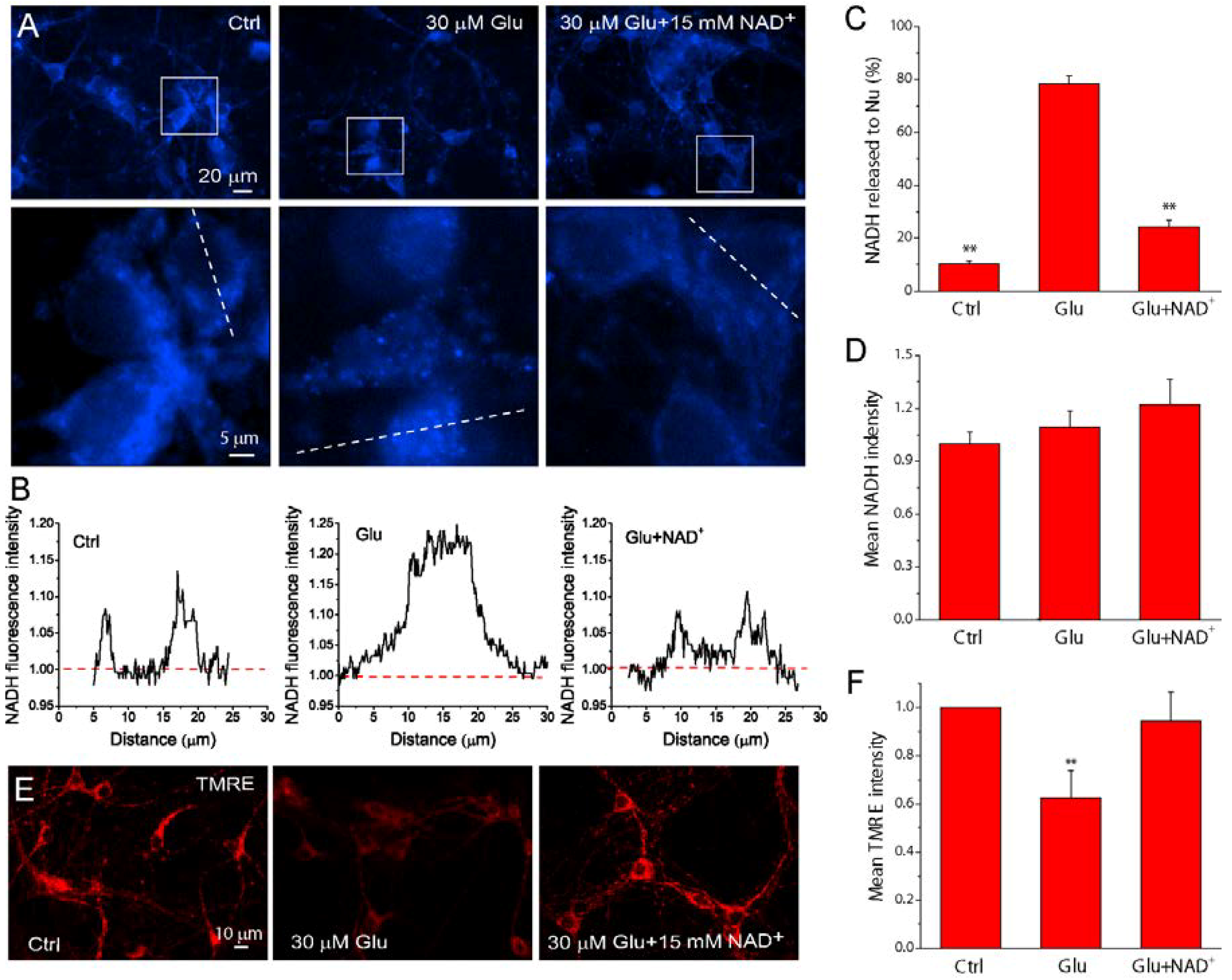
2.2. NAD+ Prevents the Translocation of AIF from Mitochondria to Nucleus


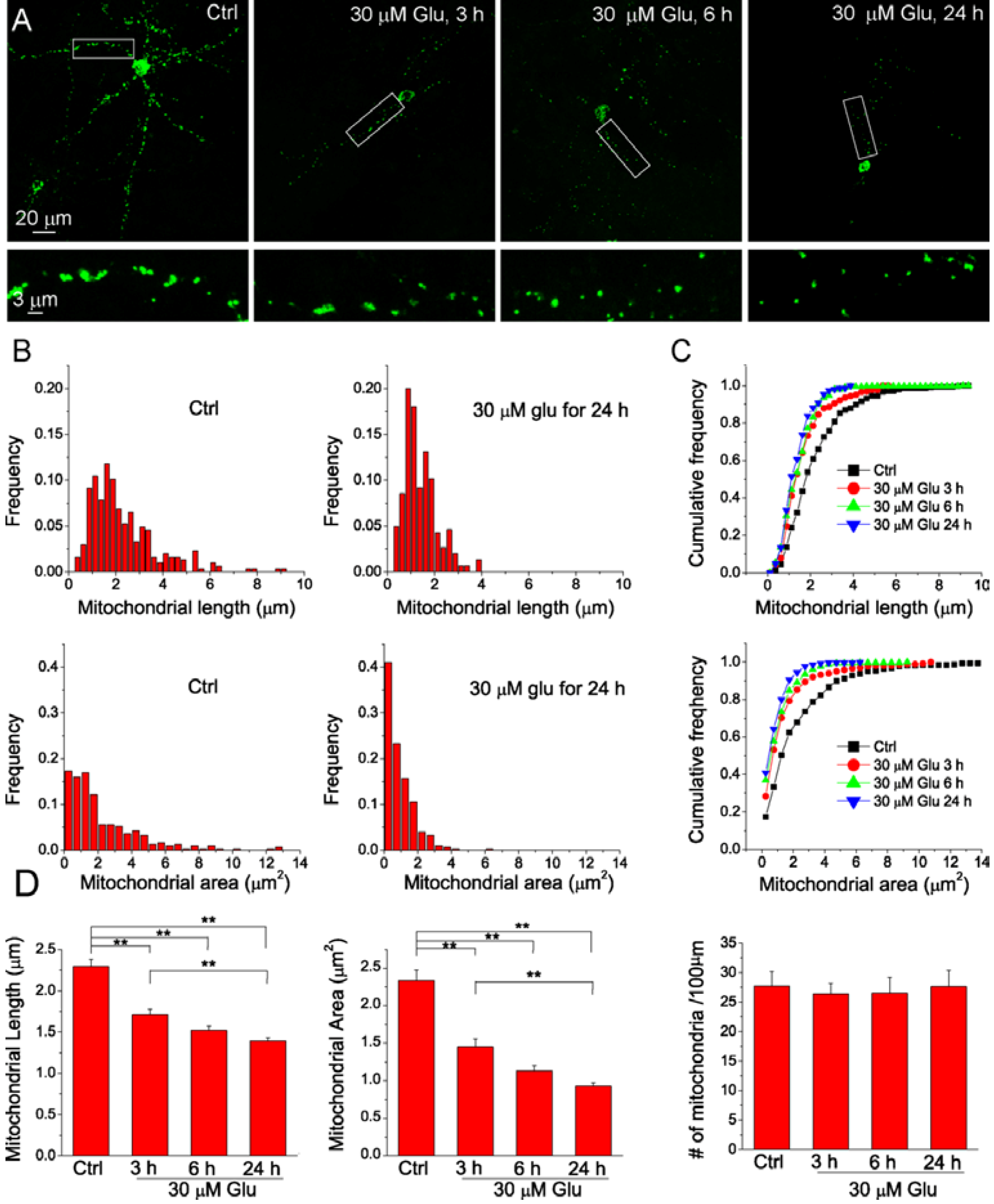
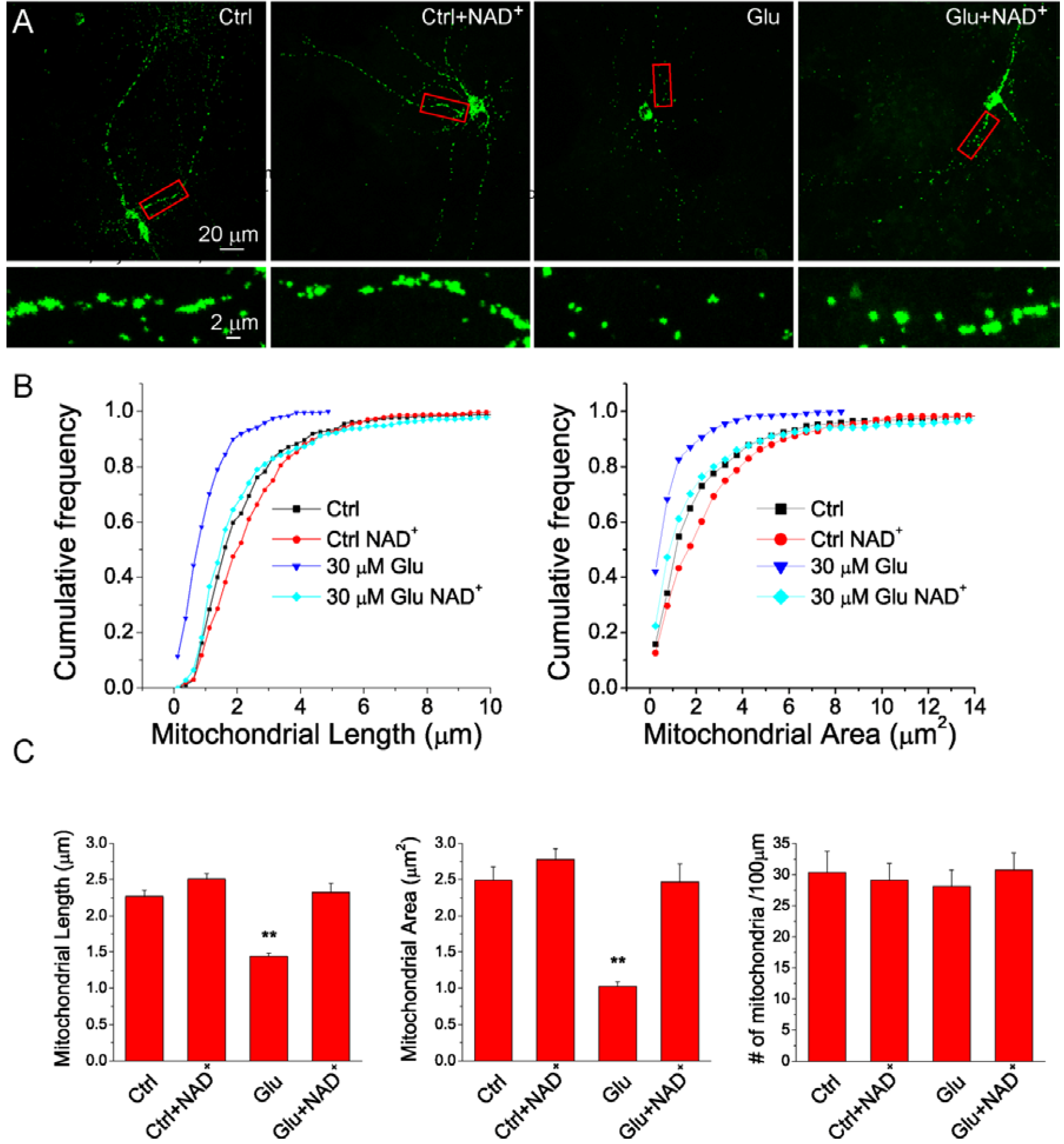
2.3. NAD+ Inhibits the Mitochondrial Fragmentation after Glutamate Excitotoxicity
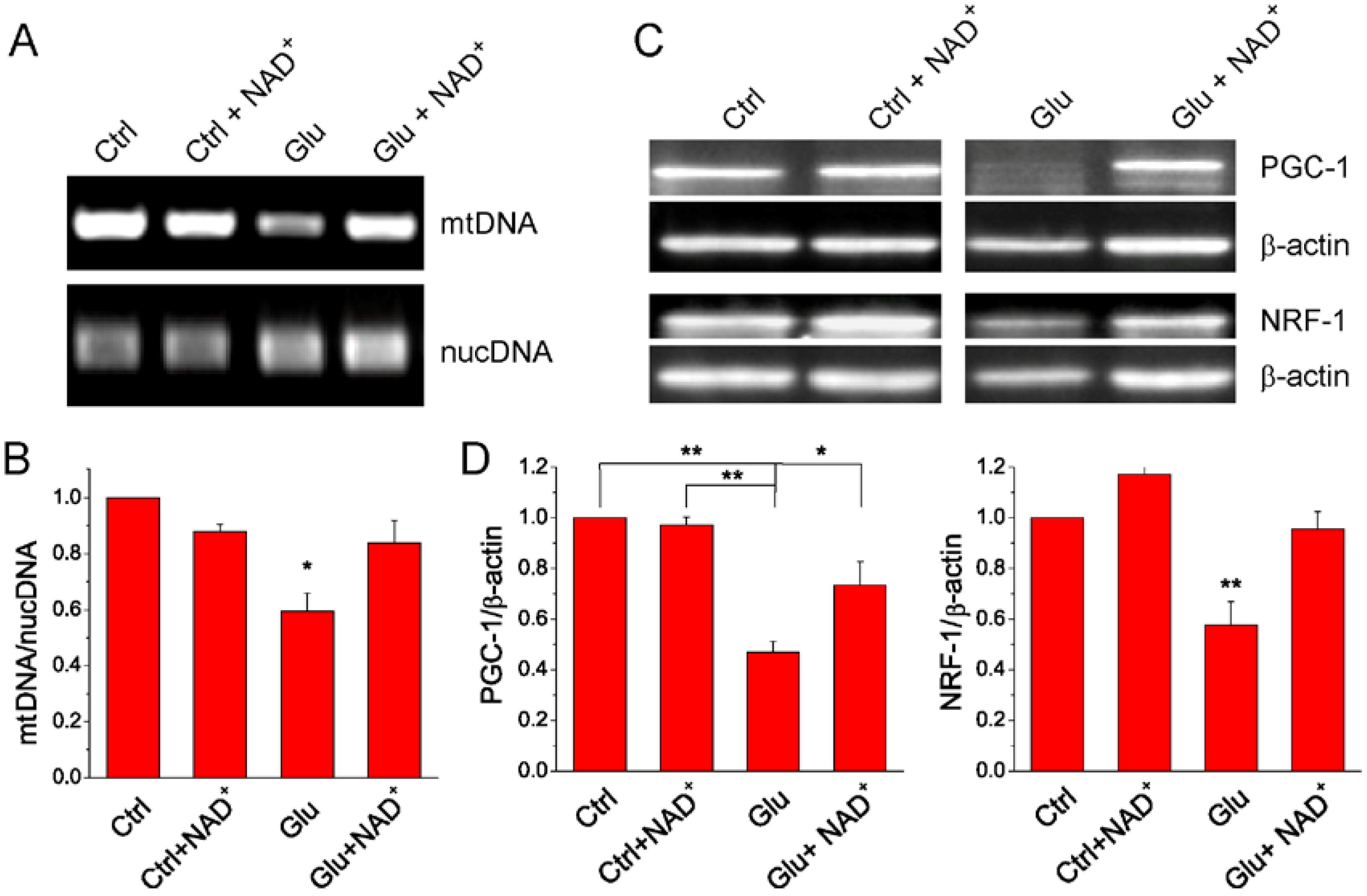
2.4. NAD+ Restores Mitochondrial Biogenesis after Glutamate Stimulation




2.5. NAD+ Prevents the Loss of Mitochondrial Integrity and Function after Glutamate Stimulation
3. Discussion
4. Materials and Methods
4.1. Primary Neuronal Cultures and in Vitro Ischemia Model
4.2. Immunofluorescent Staining
4.3. Apoptotic Neuronal Death Assay
4.4. Measurement of Mitochondrial Morphology
4.5. Assays of Mitochondrial Membrane Potential (MMP) and NADH Distribution
4.6. mtDNA Quantification
4.7. Western Blot Analysis
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Choi, D. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD+ metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Schoonjans, K.; Auwerx, J. Sirtuin functions in health and disease. Mol. Endocrinol. 2007, 21, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lavu, S.; Sinclair, D.A. Nampt/PBEF/Visfatin: a regulator of mammalian health and longevity? Exp. Gerontol. 2006, 41, 718–726. [Google Scholar]
- Lin, S.J.; Ford, E.; Haigis, M.; Liszt, G.; Guarente, L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004, 18, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Peng, I.C.; Cui, X.; Li, Y.S.; Chien, S.; Shyy, J.Y.J. Shear stress, SIRT1, and vascular homeostasis. PNAS 2010, 107, 10268–10273. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Pinto, J.T.; Ballabh, P.; Zhang, H.; Losonczy, G.; Pearson, K.J.; de Cabo, R.; Pacher, P.; Zhang, C.; et al. Resveratrol induces mitochondrial biogenesis in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H13–H20. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xing, Z.; Vosler, P.S.; Yin, H.; Li, W.; Zhang, F.; Signore, A.P.; Stetler, R.A.; Gao, Y.; Chen, J. Cellular NAD+ replenishment confers marked neuroprotection against ischemic cell death: Role of enhanced DNA repair. Stroke 2008, 39, 2587–2595. [Google Scholar] [CrossRef]
- Bi, J.; Li, H.; Ye, S.Q.; Ding, S. Pre-B-cell colony-enhancing factor exerts a neuronal protection through its enzymatic activity and the reduction of mitochondrial dysfunction in in vitro ischemic models. J. Neurochem. 2012, 120, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Han, J.; Xia, W.; Shi, S.; Liu, J.; Ying, W. NAD+ administration decreases ischemic brain damage partially by blocking autophagy in a mouse model of brain ischemia. Neurosci. Lett. 2012, 512, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pitta, M.; Mattson, M.P. Preventing NAD+ depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction. Ann. N. Y. Acad. Sci. 2008, 1147, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xie, Y.; Wang, T.; Bi, J.; Li, H.; Zhang, L.Q.; Ye, S.Q.; Ding, S. Neuronal protective role of PBEF in a mouse model of cerebral ischemia. J. Cereb. Blood Flow Metabol. 2010, 30, 1962–1971. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A. Poly(ADP-ribosyl)ation and stroke. Pharmacol. Res. 2005, 52, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Nakamura, T.; Lipton, S.A. Mitochondrial dynamics in cell death and neurodegeneration. Cell. Mol. Life Sci. 2010, 67, 3435–3447. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-b overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. PNAS 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [PubMed]
- Solenski, N.J.; diPierro, C.G.; Trimmer, P.A.; Kwan, A.L.; Helms, G.A. Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke 2002, 33, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Young, K.W.; Piñon, L.G.P.; Bampton, E.T.W.; Nicotera, P. Different pathways lead to mitochondrial fragmentation during apoptotic and excitotoxic cell death in primary neurons. J. Biochem. Mol. Toxicol. 2010, 24, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Pilon-Larose, K.; Xu, W.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Slack, R.S. The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J. Biol. Chem. 2011, 286, 4772–4782. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Bertolotti, P.; Delbarba, A.; Perego, C.; Dossena, M.; Ragni, M.; Spano, P.; Carruba, M.O.; de Simoni, M.G.; Nisoli, E. Glycogen synthase kinase-3 inhibition reduces ischemic cerebral damage, restores impaired mitochondrial biogenesis and prevents ROS production. J. Neurochem. 2011, 116, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hu, C.J.; He, Y.Y.; Yang, D.I.; Xu, J.; Hsu, C.Y. Reduction and Restoration of Mitochondrial DNA Content After Focal Cerebral Ischemia/Reperfusion. Stroke 2001, 32, 2382–2387. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Leak, R.K.; Yin, W.; Zhang, L.; Wang, S.; Gao, Y.; Chen, J. Mitochondrial biogenesis contributes to ischemic neuroprotection afforded by LPS pre-conditioning. J. Neurochem. 2012, 123 (Suppl. 2), 125–137. [Google Scholar]
- Hong, Y.; Nie, H.; Wu, D.; Wei, X.; Ding, X.; Ying, W. NAD+ treatment prevents rotenone-induced apoptosis and necrosis of differentiated PC12 cells. Neurosci. Lett. 2014, 2014, 46–50. [Google Scholar] [CrossRef]
- Pittelli, M.; Felici, R.; Pitozzi, V.; Giovannelli, L.; Bigagli, E.; Cialdai, F.; Romano, G.; Moroni, F.; Chiarugi, A. Pharmacological effects of exogenous NAD+ on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 2011, 80, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Eliasson, M.J.L.; Sampei, K.; Mandir, A.S.; Hurn, P.D.; Traystman, R.J.; Bao, J.; Pieper, A.; Wang, Z.Q.; Dawson, T.M.; Snyder, S.H.; et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Wang, Z.Q.; Namura, S.; Waeber, C.; Moskowitz, M.A. Ischemic brain injury is mediated by the activation of poly(ADP-Ribose)polymerase. J. Cereb. Blood Flow Metab. 1997, 17, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, S.W.; Koh, D.W.; Lew, J.; Coombs, C.; Bowers, W.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci. 2004, 24, 10963–10973. [Google Scholar] [CrossRef] [PubMed]
- Niimura, M.; Takagi, N.; Takagi, K.; Mizutani, R.; Ishihara, N.; Matsumoto, K.; Funakoshi, H.; Nakamura, T.; Takeo, S. Prevention of apoptosis-inducing factor translocation is a possible mechanism for protective effects of hepatocyte growth factor against neuronal cell death in the hippocampus after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2006, 26, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Bhavnani, B.R. Glutamate-induced apoptosis in primary cortical neurons is inhibited by equine estrogens via down-regulation of caspase-3 and prevention of mitochondrial cytochrome c release. BMC Neurosci. 2005, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, D.; Qi, W.; Yu, D.; Jiang, H.; Han, C.; Kim, M.J.; Katsuno, K.; Hsieh, Y.H.; Miyakawa, T.; Salvi, R.; et al. Addition of exogenous NAD+ prevents mefloquine-induced neuroaxonal and hair cell degeneration through reduction of caspase-3-mediated apoptosis in cochlear organotypic cultures. PLoS One 2013, 8, e79817. [Google Scholar] [CrossRef] [PubMed]
- Rintoul, G.L.; Filiano, A.J.; Brocard, J.B.; Kress, G.J.; Reynolds, I.J. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci. 2003, 23, 78881–78888. [Google Scholar]
- Rube, D.A.; van der Bliek, A.M. Mitochondrial morphology is dynamic and varied. Mol. Cell. Biochem. 2004, 2004, 256–257. [Google Scholar]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Berman, S.B.; Chen, Y.; Qi, B; McCaffery, J.M.; Rucker, E.B., III; Goebbels, S.; Nave, K.; Arnold, B.A.; Jonas, E.A.; Pineda, F.J.; et al. Bcl-xL increases mitochondrial fission, fusion, and biomass in neurons. J. Cell Biol. 2009, 184, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhu, W.; Jia, J.; Zhang, C.; Xu, Y. NMDA receptor dependent PGC-1alpha up-regulation protects the cortical neuron against oxygen-glucose deprivation/reperfusion injury. J. Mol. Neurosci. 2009, 39, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Ramadori, G.; Lee, C.E.; Bookout, A.L.; Lee, S.; Williams, K.W.; Anderson, J.; Elmquist, J.K.; Coppari, R. Brain SIRT1: Anatomical distribution and regulation by energy availability. J. Neurosci. 2008, 28, 9989–96. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.R.; Wang, Z.Y.; Zhu, X.L.; Wu, X.X.; Li, E.G.; Xu, Y. Icariin protects against brain injury by enhancing SIRT1-dependent PGC-1α expression in experimental stroke. Neuropharmacology 2010, 59, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, F.; Carcenac, C.; Gonthier, B.; Cottet-Rousselle, C.; Chauvin, C.; Barret, L.; Leverve, X.; Savasta, M.; Fontaine, E. Mitochondrial permeability transition pore inhibitors prevent ethanol-induced neuronal death in mice. Chem. Res. Toxicol. 2013, 26, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Signore, A.P.; Iwai, M.; Cao, G.; Gao, Y.; Chen, J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 2008, 39, 3057–3063. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Li, H.; Ding, S. The Effects of NAD+ on Apoptotic Neuronal Death and Mitochondrial Biogenesis and Function after Glutamate Excitotoxicity. Int. J. Mol. Sci. 2014, 15, 20449-20468. https://doi.org/10.3390/ijms151120449
Wang X, Li H, Ding S. The Effects of NAD+ on Apoptotic Neuronal Death and Mitochondrial Biogenesis and Function after Glutamate Excitotoxicity. International Journal of Molecular Sciences. 2014; 15(11):20449-20468. https://doi.org/10.3390/ijms151120449
Chicago/Turabian StyleWang, Xiaowan, Hailong Li, and Shinghua Ding. 2014. "The Effects of NAD+ on Apoptotic Neuronal Death and Mitochondrial Biogenesis and Function after Glutamate Excitotoxicity" International Journal of Molecular Sciences 15, no. 11: 20449-20468. https://doi.org/10.3390/ijms151120449