Towards Controlling the Glycoform: A Model Framework Linking Extracellular Metabolites to Antibody Glycosylation


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
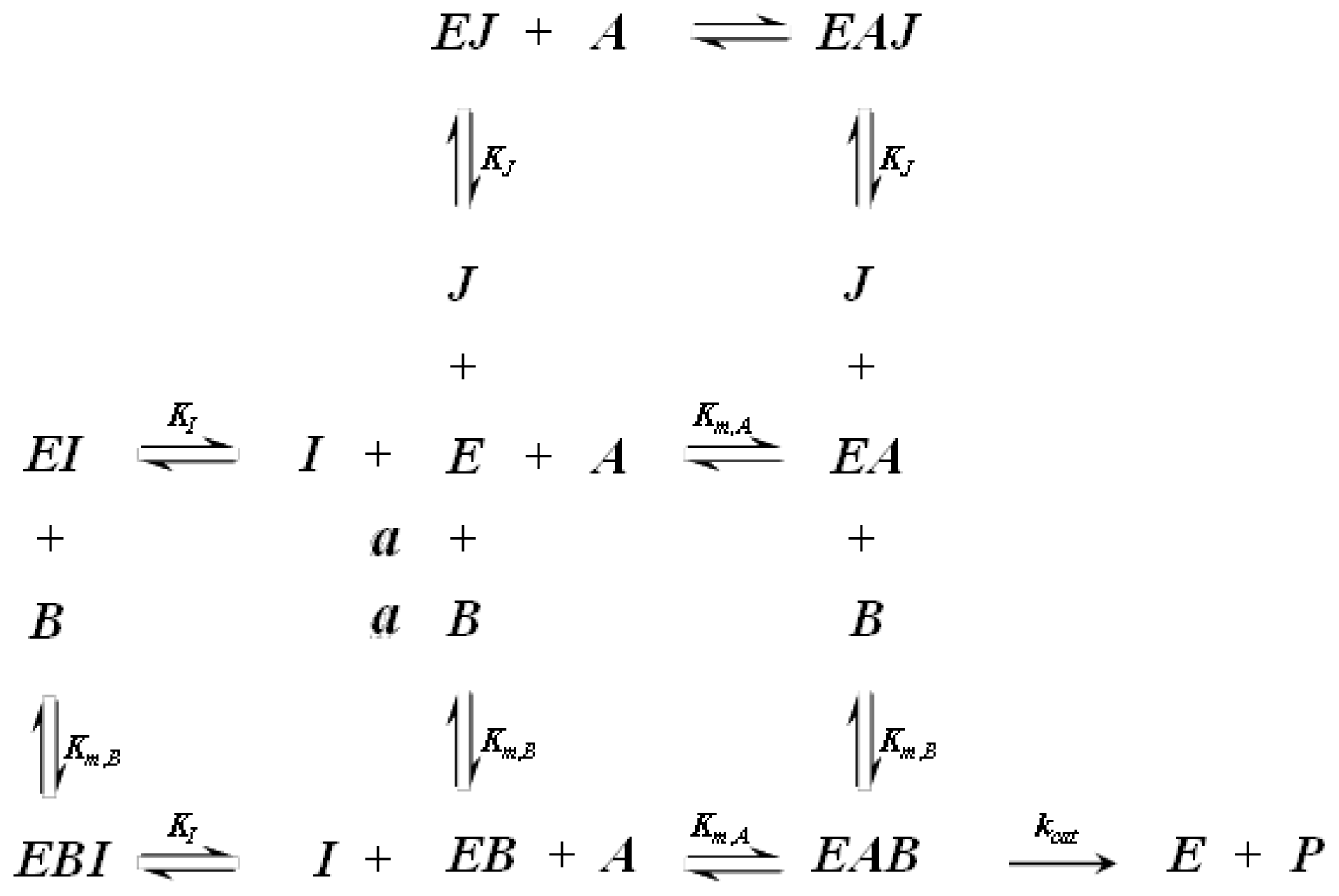{kind=link}
| V | culture volume | L |
| XV | cell density | cells/L |
| t | time | h |
| μ | cell growth rate | h−1 |
| kd | cell death rate | h−1 |
| Fout | flow rate out of culture | L/h |
| Glcext | extracellular glucose concentration | mM |
| Glnext | extracellular glutamine concentration | mM |
| Fin | flow rate into culture | L/h |
| Glcfeed | feed glucose concentration | mM |
| KA | activator species saturation coefficient | mM |
| KM | species saturation coefficient | mM |
| kd,max | maximum cell death rate | h−1 |
| Kd | species depletion coefficient | mM |
| q | species cellular production | mmol/(h-cell) |
| Y | species yield | cell/mmol |
| m | species cell maintenance term | mmol/(h-cell) |
| mAb | Antibody product titer | mM |
| Nuc | Intracellular nucleotide concentration | mM |
| KTP | Transport protein species saturation coefficient | mM |
| Vcell | cell volume | L |
| DNAf | nucleotide fraction in DNA | dimensionless |
| mDNA | cellular DNA mass | mg/cell |
| mRNA | cellular RNA mass | mg/cell |
| Mr | molecular species mass | mg/mmol |
| RNAf | nucleotide fraction in RNA | dimensionless |
| kcat | enzyme turnover rate | h−1 |
| Ki | Species inhibition constant | mM |
| E0 | Initial enzyme concentration | mM |
| Ngly,cell | Number of glycans per cell | mmol/cell |
| NNSD,gly | NSDs consumed per host cell glycan | mmol/mmol |
| Ngly,mAb | Number of glycans per antibody | mmol/mmol |
| NNSD,mAb | NSDs consumed per antibody | mmol/mmol |
| FmAb | Antibody production rate | mmol/h |
1. Introduction
Current Problems Resulting from Glycans and Causes of Variation
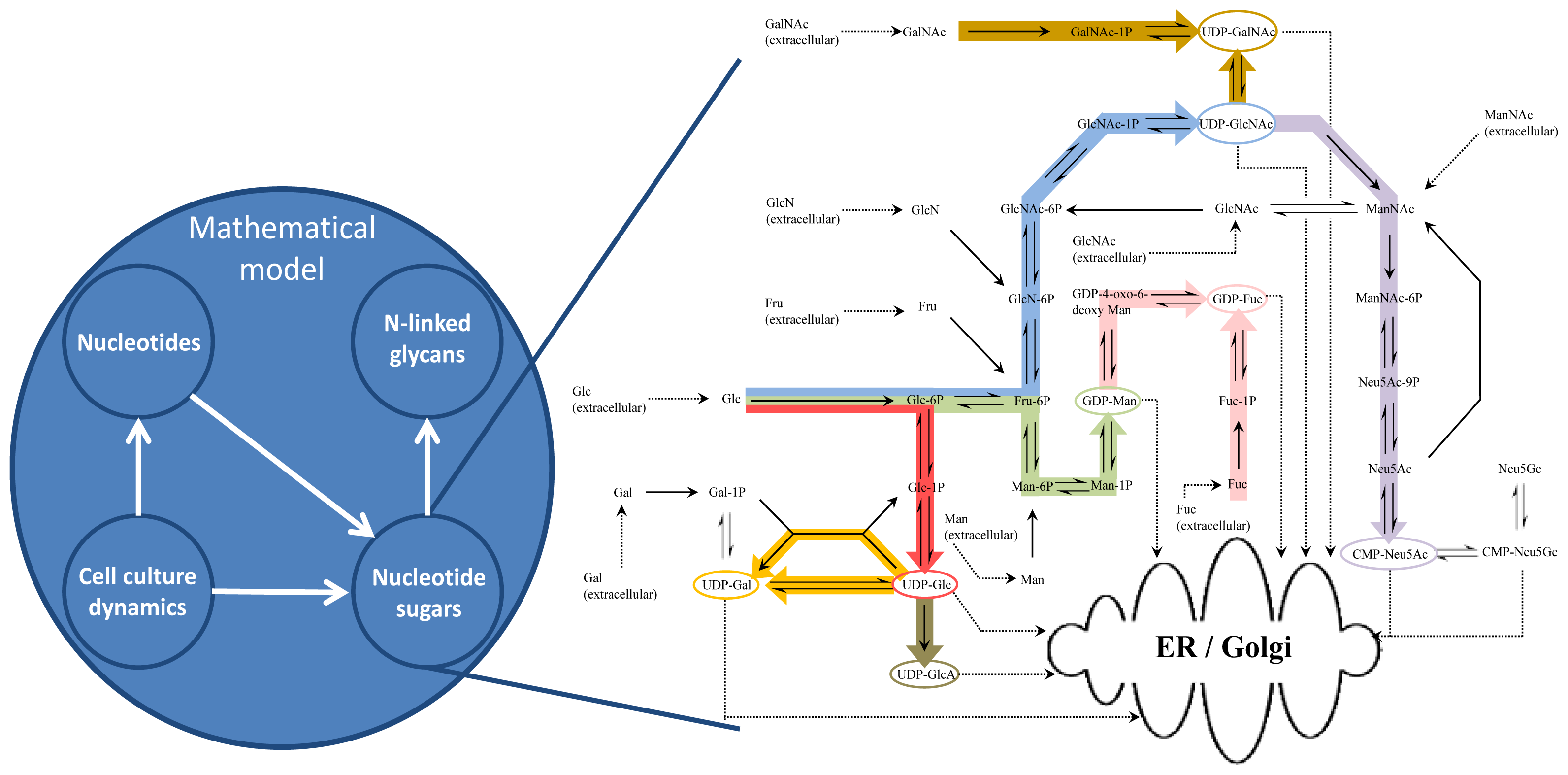
2. Mathematical Model Development
2.1. Cell Culture Dynamics Model
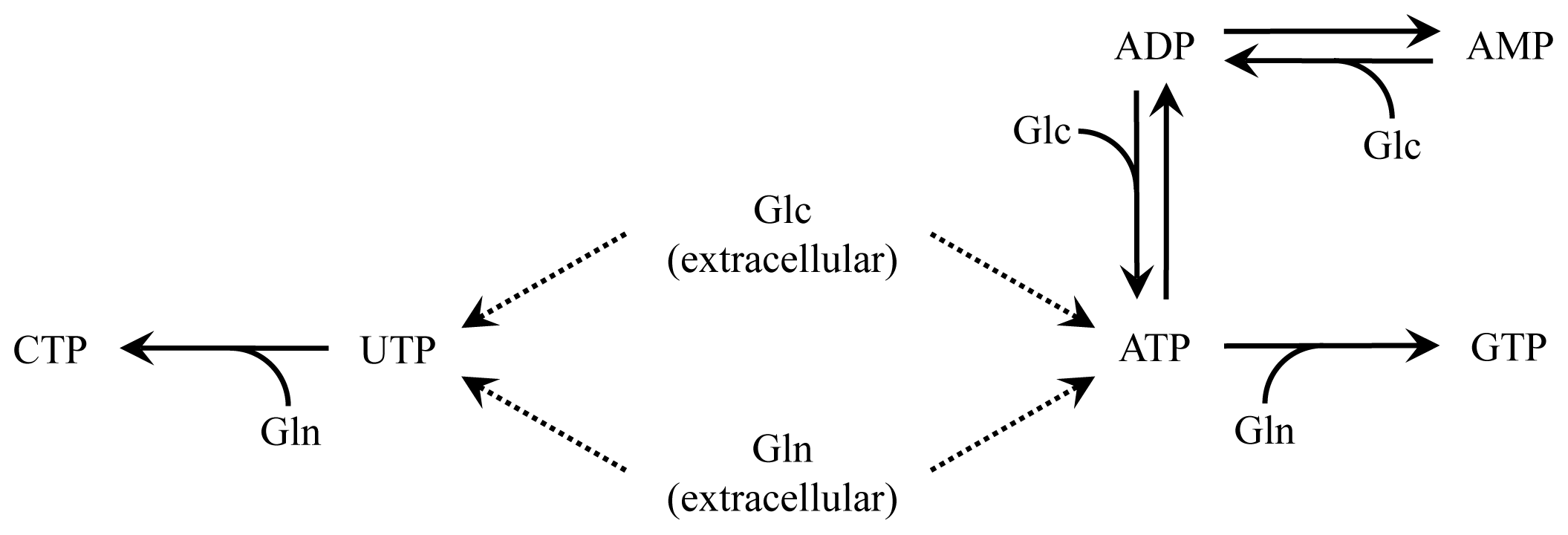
2.2. Nucleotide Model
2.3. Nucleotide Sugar Synthesis Model
- Equilibrium is rapidly reached for all intermediate reactants;
- Rate-limiting steps are irreversible (Table S1);
- Where water is required for catalysis, full enzyme saturation is assumed due to the aqueous environment of the cytoplasm;
- Where more than one substrate is required for catalysis, a random order of substrate binding is assumed, unless reported otherwise;
- Rapid dissociation of reaction products from enzyme;
- Michaelis-Menten kinetics are assumed to hold true, unless reported otherwise;
- All enzyme and transport protein concentrations throughout the network are constant.
2.4. Parameter Estimation
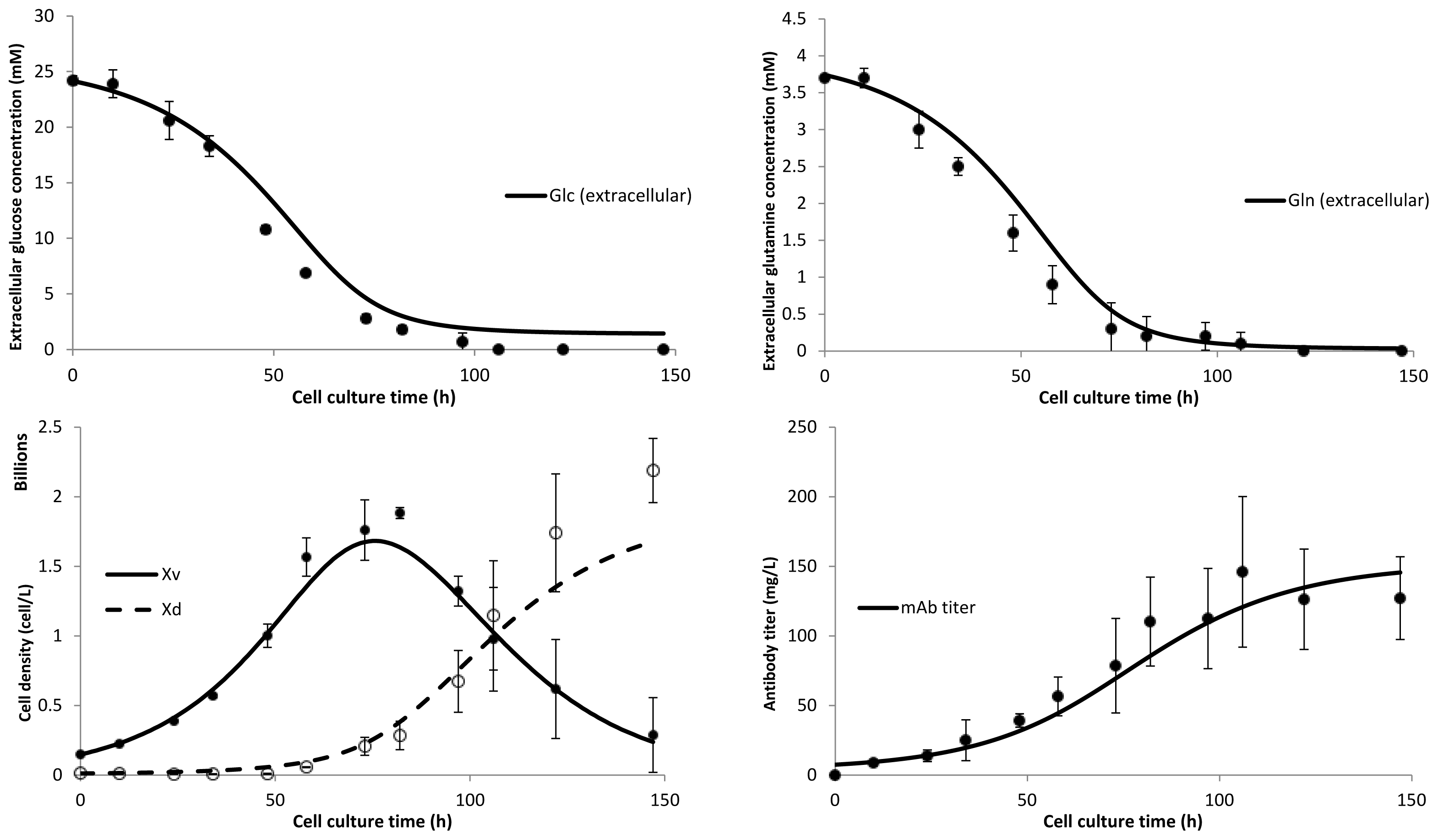
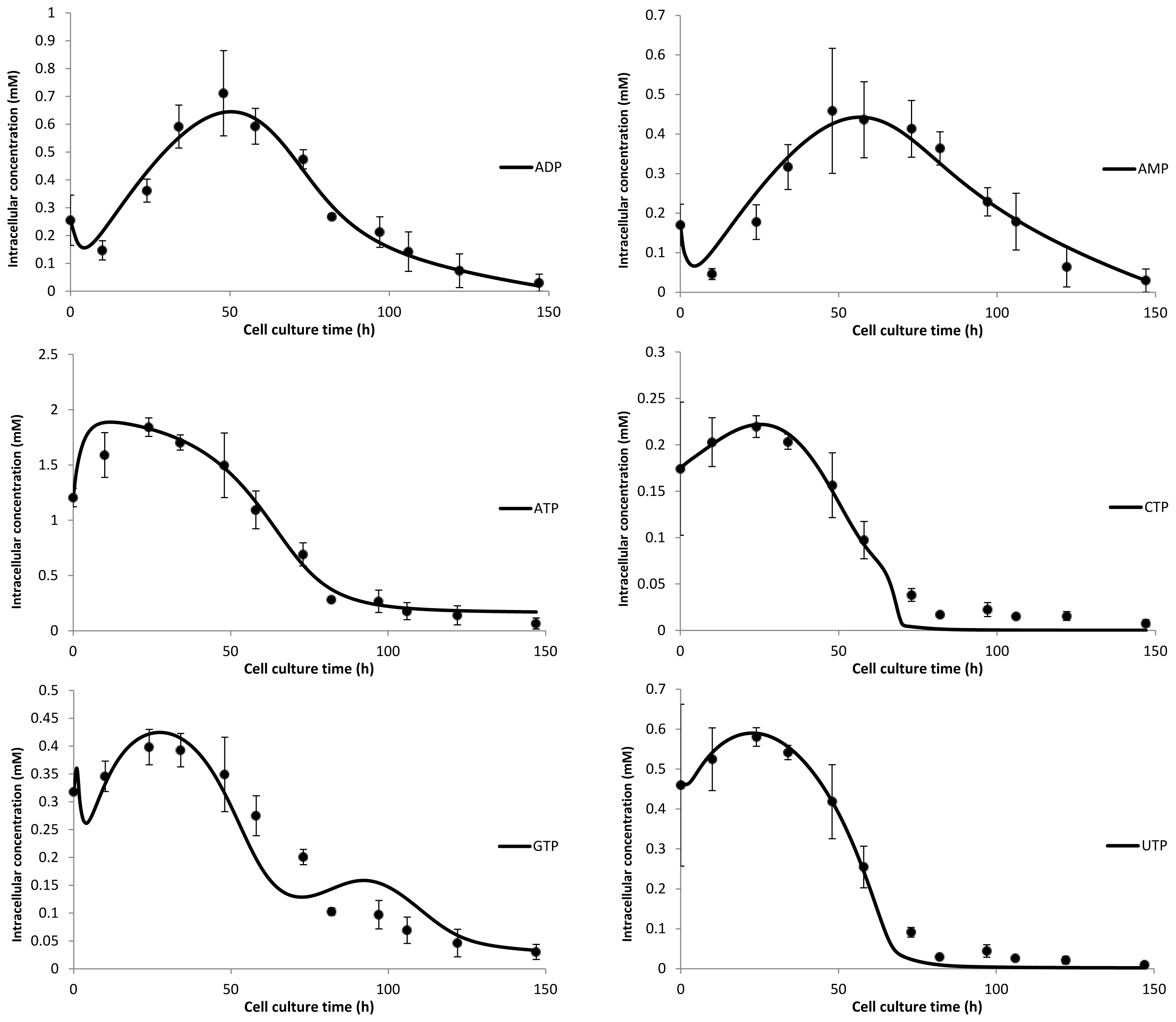
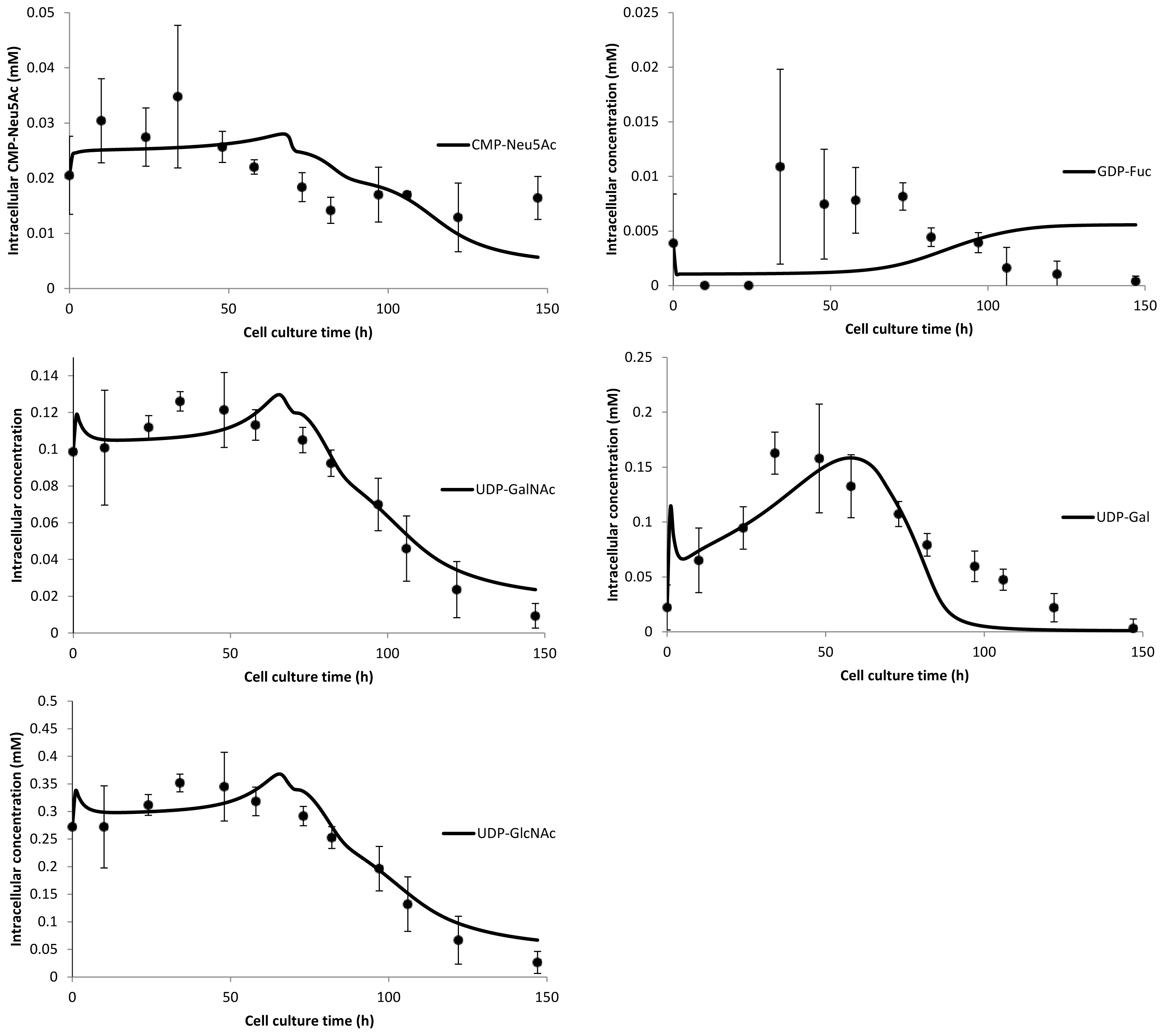
3. Model Performance and Discussion
4. Materials and Methods
4.1. Cell Culture, Metabolite Monitoring and Antibody Quantification
4.2. Intracellular Nucleotide and Nucleotide Sugar Extraction
4.3. Characterization of Intracellular Nucleotides and Nucleotide Sugars
4.4. Glycan Purification and Analysis
5. Conclusions
Acknowledgments
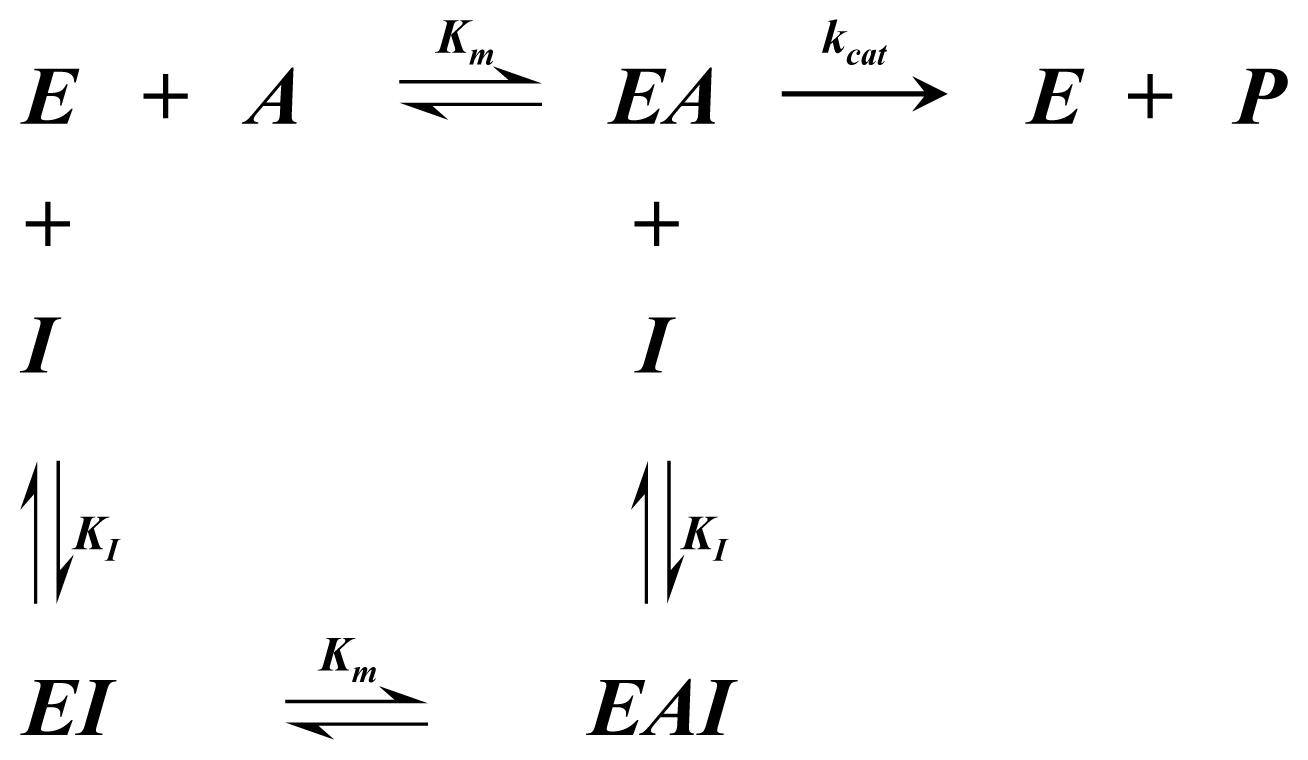
Appendix 1—Enzyme Mechanisms
A1.1. Single Substrate Michaelis-Menten Rate Equation

| Equation (1) Rate of reaction for Single substrate Michaelis-Menten kinetics | |
| Equation (2) Rate of reaction for Single substrate Michaelis-Menten kinetics including competitive inhibition of species A | |
| Equation (3) Rate of reaction for Single substrate Michaelis-Menten kinetics including non-competitive inhibition |
A1.2. Random Order Bi-Bi Kinetics

| Equation (4) Rate of reaction for random order bi-bi kinetics | |
| Equation (5) Rate of reaction for random order bi-bi kinetics including competitive inhibition of species A and B | |
| Equation (6) Rate of reaction for random order bi-bi kinetics including non-competitive inhibition | |
A1.3. Ordered Bi-Bi Kinetics

| Equation (7) Rate of reaction for ordered bi-bi kinetics | |
| Equation (8) Rate of reaction for ordered bi-bi kinetics including competitive inhibition of species A and B | |
| Equation (9) Rate of reaction for ordered bi-bi kinetics including non-competitive inhibition |
A1.4. Ping-Pong Bi-Bi Kinetics

| Equation (10) Rate of reaction for ping-pong bi-bi kinetics | |
| Equation (11) Rate of reaction for ping-pong bi-bi kinetics including competitive inhibition of species A and B | |
| Equation (12) Rate of reaction for ping-pong bi-bi kinetics including non-competitive inhibition |
A1.5. Ping-Pong Ter-Ter Kinetics

| Equation (13) Rate of reaction for ping-pong ter-ter kinetics including competitive inhibition of species A |
A1.5. Hexokinase Rate of Reaction Expression

| Equation (14) Rate of reaction for hexokinase based on the above reaction scheme |
A1.6. Glycolysis
A1.7. Hill Coefficients
Appendix 2—Relative Abundances of Activated Human B-Cell Glycans
| Glycan structure | Sugar frequency per glycan (mol/mol) | |||||
|---|---|---|---|---|---|---|
| Species abundance (%) | GlcNAc | Man | Gal | Fuc | CMP-Neu5Ac | |
 | 7.37 | 2 | 5 | |||
 | 10.86 | 2 | 6 | |||
 | 1.75 | 4 | 3 | 1 | ||
 | 13.40 | 2 | 7 | |||
 | 1.75 | 4 | 3 | 1 | 1 | |
 | 14.85 | 2 | 8 | |||
 | 0.69 | 4 | 3 | 2 | 1 | |
 | 0.47 | 5 | 3 | 1 | 1 | |
 | 17.69 | 2 | 9 | |||
 | 1.87 | 4 | 3 | 2 | 1 | |
 | 0.44 | 5 | 3 | 2 | 1 | |
 | 2.97 | 4 | 3 | 2 | 1 | 1 |
 | 0.49 | 4 | 3 | 2 | 2 | 1 |
 | 1.23 | 4 | 3 | 2 | 2 | |
 | 10.78 | 4 | 3 | 2 | 1 | 2 |
 | 2.59 | 5 | 3 | 3 | 1 | 1 |
 | 5.03 | 5 | 3 | 2 | 1 | 2 |
 | 0.34 | 6 | 3 | 3 | 1 | 1 |
 | 1.35 | 5 | 3 | 3 | 1 | 2 |
 | 0.59 | 6 | 3 | 4 | 1 | 1 |
 | 0.12 | 6 | 3 | 3 | 1 | 2 |
 | 0.10 | 7 | 3 | 4 | 1 | 1 |
 | 0.31 | 5 | 3 | 3 | 1 | 3 |
 | 0.68 | 6 | 3 | 4 | 1 | 2 |
 | 0.10 | 7 | 3 | 5 | 1 | 1 |
 | 0.09 | 7 | 3 | 4 | 1 | 2 |
 | 0.18 | 6 | 3 | 4 | 1 | 3 |
 | 0.36 | 7 | 3 | 5 | 1 | 2 |
 | 0.09 | 7 | 3 | 5 | 1 | 3 |
 | 0.09 | 8 | 3 | 6 | 1 | 2 |
| Glycan structure | Sugar frequency per glycan (mol/mol) | ||||
|---|---|---|---|---|---|
| Species abundance (%) | GlcNAc | GalNAc | Gal | CMP-Neu5Ac | |
 | 30.34 | 1 | 1 | 1 | |
 | 2.18 | 1 | 1 | 2 | |
 | 53.64 | 1 | 1 | 2 | |
 | 11.42 | 1 | 1 | 2 | 1 |
 | 0.77 | 1 | 1 | 3 | |
 | 1.27 | 1 | 1 | 2 | 2 |
 | 0.38 | 2 | 1 | 3 | 1 |
Appendix 3—Estimated Parameter Values and Non-Nucleotide Species
A3.1. NSD Metabolic Network Parameter Values
| Parameter name | Parameter value |
|---|---|
| E15a | 2.5400E−05 |
| E8a | 0.0000E+00 |
| E26b | 0.0000E+00 |
| E28a | 0.0000E+00 |
| E34a | 0.0000E+00 |
| E19a | 6.4400E−06 |
| E21a | 6.8586E−06 |
| Eglyc | 7.0800E−05 |
| Ki15a_GDPFuc | 2.1200E−04 |
| E29a | 1.3313E−03 |
| E1c | 1.5096E−03 |
| E1a | 1.8477E−03 |
| Km17a_Man6P | 2.5300E−03 |
| E23a | 3.7138E−03 |
| E12a | 3.9000E−03 |
| E32a | 3.9000E−03 |
| E14a | 3.9000E−03 |
| E16a | 3.9000E−03 |
| E20a | 3.9000E−03 |
| E2a | 3.9000E−03 |
| E37a | 3.9000E−03 |
| E3b | 3.9000E−03 |
| E40a | 3.9000E−03 |
| E4a | 3.9000E−03 |
| E4b | 3.9000E−03 |
| E17a | 3.9532E−03 |
| E22a | 1.0000E−02 |
| E3a | 1.4167E−02 |
| E38a | 3.5128E−02 |
| E13a | 3.9000E−02 |
| E31a | 3.9000E−02 |
| E33a | 3.9000E−02 |
| E5a | 3.9000E−02 |
| E6a | 3.9000E−02 |
| Km26a_Glc6P | 9.3842E−02 |
| Km22a_UDPGlc | 2.0697E−01 |
| Km7b_UDPGlcNAc | 8.2129E−01 |
| Gln_coef | 1.0000E+00 |
| n29a_CMPNeu5Ac | 4.2000E+00 |
| Ki_SA_Tra_UDPGlcNAc | 6.8885E+00 |
| Km19a_Fru6P | 1.9700E+01 |
| k5aB | 6.0000E+02 |
| Ki34a_CMPNeu5Gc | 1.0000E+03 |
| k26aF | 2.0972E+03 |
| k6aB | 2.3900E+04 |
| k17aF | 7.0928E+04 |
| k22aB | 1.6151E+05 |
| Ki29a_CMPNeu5Ac | 5.2433E+05 |
| k21aB | 4.2900E+06 |
| k21aF | 6.1200E+06 |
| k19aF | 1.5900E+07 |
| k27aF | 1.1200E+08 |
| Parameter name | Parameter value |
|---|---|
| k_T_gln | 3.3800E−06 |
| Kd_Glc_ext | 1.0051E−01 |
| Kd_Gln_ext | 1.1872E−02 |
| Km_Glc_ext | 2.6700E+00 |
| Km_Gln_ext | 1.2000E+00 |
| mu_d_max_glc | 3.9300E−01 |
| mu_d_max_gln | 6.2053E−02 |
| mu_g_max | 6.6745E−02 |
| Y_ext_glc | 9.1600E+07 |
| Y_ext_gln | 5.6400E+08 |
| Y_mAb_mu | 0.0000E+00 |
| Y_mAb_Xv | 1.1400E−09 |
| Glc_in | 0.0000E+00 |
| Gln_in | 0.0000E+00 |
| F_in | 0.0000E+00 |
| F_out | 0.0000E+00 |
| Parameter name | Parameter value |
|---|---|
| Kdf_10_Gln | 2.6356E+00 |
| Kdf_11_Glc | 2.1296E+00 |
| Kdf_11_Gln | 1.3425E+00 |
| Kdf_12_ATP | 1.1302E+01 |
| Kdf_13_ADP | 3.9892E−04 |
| Kdf_13_Glc | 4.7469E+00 |
| Kdf_14_ADP | 2.5000E+01 |
| Kdf_15_AMP | 1.1312E+01 |
| Kdf_15_Glc | 2.3025E+00 |
| Kdf_8_Glc | 1.2067E+00 |
| Kdf_8_Gln | 2.4832E+00 |
| Kdf_9_Gln | 2.1804E+00 |
| Kdf_9_UTP | 5.0374E−03 |
| Kdout_ATP | 1.0000E−03 |
| Kdout_CTP | 1.0000E−03 |
| Kdout_GTP | 1.0000E−03 |
| Kdout_UTP | 1.0000E−03 |
| kf_10 | 7.6621E+00 |
| kf_11 | 6.7500E+00 |
| kf_12 | 3.2344E+00 |
| kf_13 | 2.1454E−01 |
| kf_14 | 1.1649E+02 |
| kf_15 | 8.6900E+01 |
| kf_8 | 7.9486E+00 |
| kf_9 | 1.3393E+00 |
| Parameter name | Parameter value |
|---|---|
| KdiFucTA | 0.0000E+00 |
| KdiFucTB | 0.0000E+00 |
| KdiGalTa1A | 1.0709E+02 |
| KdiGalTa1B | 7.2051E+00 |
| KdiGalTa2A | 3.4573E+01 |
| KdiGntII | 7.7067E+01 |
A3.2. Non-NSD Species
| Intracellular species | Intracellular conc. (mM) | Source tissue |
|---|---|---|
| Acetyl Coenzyme A (ACoA) | 0.029 | Rat liver |
| Coenzyme A (CoA) | 0.13 | Rat liver |
| Glucose-1,6-biphosphate (Glc16PP) | 0.014 | Mouse liver |
| Nicotinamide adenine dinucleotide (NAD) | 0.76 | Rat liver |
| Nictotinamide adenine dinucleotide phosphate (NADP) | 0.067 | Rat liver |
| Nictotinamide adenine dinucleotide phosphate, reduced (NADPH) | 0.30 | Rat liver |
| Phosphoenolypyruvic acid (PEP) | 0.11 | Mouse liver |
| Inorganic phosphate (PPi) | 3.37 | Rat liver |
| Pyruvate (Pyr) | 0.18 | Mouse liver |
Conflicts of Interest
References
- Evaluate Pharma Embracing the Patent Cliff. In World Preview 2018; EvaluatePharma Ltd.: London, UK, 2013; p. 38.
- Kyriakopoulos, S.; Kontoravdi, C. Analysis of the landscape of biologically-derived pharmaceuticals in Europe: Dominant production systems molecule types on the rise and approval trends. Eur. J. Pharm. Sci. 2013, 48, 428–441. [Google Scholar]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.A.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fc gamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar]
- Matsumiya, S.; Yamaguchi, Y.; Saito, J.; Nagano, M.; Sasakawa, H.; Otaki, S.; Satoh, M.; Shitara, K.; Kato, K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J. Mol. Biol. 2007, 368, 767–779. [Google Scholar]
- Hodoniczky, J.; Zheng, Y.Z.; James, D.C. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol. Progr. 2005, 21, 1644–1652. [Google Scholar]
- Anthony, R.M.; Nimmerjahn, F.; Ashline, D.J.; Reinhold, V.N.; Paulson, J.C.; Ravetch, J.V. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG fc. Science 2008, 320, 373–376. [Google Scholar]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.Q.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar]
- Mullard, A. Can next-generation antibodies offset biosimilar competition? Nat. Rev. Drug Discov. 2012, 11, 426–428. [Google Scholar]
- Jedrzejewski, P.M.; Jimenez del Val, I.; Polizzi, K.M.; Kontoravdi, C. Applying quality by design to glycoprotein therapeutics: Experimental and computational efforts of process control. Pharm. Bioprocess. 2013, 1, 51–69. [Google Scholar]
- Balaquer, E.; Neususs, C. Intact glycoform characterization of erythropoietin-alpha and erythropoietin-beta by CZE-ESI-TOF-MS. Chromatographia 2006, 64, 351–357. [Google Scholar]
- Krambeck, F.J.; Bennun, S.V.; Narang, S.; Choi, S.; Yarema, K.J.; Betenbaugh, M.J. A mathematical model to derive N-glycan structures and cellular enzyme activities from mass spectrometric data. Glycobiology 2009, 19, 1163–1175. [Google Scholar]
- Del Val, I.J.; Kontoravdi, C.; Nagy, J.M. Towards the implementation of quality by design to the production of therapeutic monoclonal antibodies with desired glycosylation patterns. Biotechnol. Progr. 2010, 26, 1505–1527. [Google Scholar]
- Hills, A.E.; Patel, A.; Boyd, P.; James, D.C. Metabolic control of recombinant monoclonal antibody N-glycosylation in GS-NS0 cells. Biotechnol. Bioeng. 2001, 75, 239–251. [Google Scholar]
- Grainger, R.K.; James, D.C. CHO cell line specific prediction and control of recombinant monoclonal antibody N-glycosylation. Biotechnol. Bioeng. 2013, 110, 2970–2983. [Google Scholar]
- Wong, N.S.C.; Wati, L.; Nissom, P.M.; Feng, H.T.; Lee, M.M.; Yap, M.G.S. An investigation of intracellular glycosylation activities in CHO Cells: Effects of nucleotide sugar precursor feeding. Biotechnol. Bioeng. 2010, 107, 321–336. [Google Scholar]
- Rathore, A.S.; Winkle, H. Quality by design for biopharmaceuticals. Nat. Biotechnol. 2009, 27, 26–34. [Google Scholar]
- del Val, I.J.; Nagy, J.M.; Kontoravdi, C. A dynamic mathematical model for monoclonal antibody N-linked glycosylation and nucleotide sugar donor transport within a maturing Golgi apparatus. Biotechnol. Progr. 2011, 27, 1730–1743. [Google Scholar]
- gPROMS Introductory User Guideversion—Release 3.5.0; Process Systems Enterprise, Ltd.: London, UK, 2011.
- Xie, L.Z.; Wang, D.I.C. Stoichiometric analysis of animal-cell growth and its application in medium design. Biotechnol. Bioeng. 1994, 43, 1164–1174. [Google Scholar]
- Nolan, R.P.; Lee, K. Dynamic model of CHO cell metabolism. Metab. Eng. 2011, 13, 108–124. [Google Scholar]
- Bonarius, H.P.; Hatzimanikatis, V.; Meesters, K.P.; de Gooijer, C.D.; Schmid, G.; Tramper, J. Metabolic flux analysis of hybridoma cells in different culture media using mass balances. Biotechnol. Bioeng. 1996, 50, 299–318. [Google Scholar]
- KEGG (Kyoto Encylopedia of Genes and Genomes). Available online: http://www.genome.jp/kegg/ (accessed on 10 March 2014).
- BRENDA (BRaunschweig ENzyme DAtabase). Available online: http://www.brenda-enzymes.org (accessed on 10 March 2014).
- Springer, M.S.; Stanhope, M.J.; Madsen, O.; de Jong, W.W. Molecules consolidate the placental mammal tree. Trends Ecol. Evol. 2004, 19, 430–438. [Google Scholar]
- Rijcken, W.R.P.; Overdijk, B.; Vandeneijnden, D.H.; Ferwerda, W. The effect of increasing nucleotide sugar concentrations on the incorporation of sugars into glycoconjugates in rat hepatocytes. Biochem. J. 1995, 305, 865–870. [Google Scholar]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation as deduced from analysis of the SWISS-PROT database. Bba-Gen. Subj. 1999, 1473, 4–8. [Google Scholar]
- Chen, N.; Koumpouras, G.C.; Polizzi, K.M.; Kontoravdi, C. Genome-based kinetic modeling of cytosolic glucose metabolism in industrially relevant cell lines: Saccharomyces cerevisiae and Chinese hamster ovary cells. Bioproc. Biosyst. Eng. 2012, 35, 1023–1033. [Google Scholar]
- Wong, D.C.F.; Wong, N.S.C.; Goh, J.S.Y.; May, L.M.; Yap, M.G.S. Profiling of N-glycosylation gene expression in CHO cell fed-batch cultures. Biotechnol. Bioeng. 2010, 107, 516–528. [Google Scholar]
- Frame, K.K.; Hu, W.S. Cell-Volume Measurement as an estimation of mammalian-cell biomass. Biotechnol. Bioeng. 1990, 36, 191–197. [Google Scholar]
- Lee, M.S.; Lee, G.M. Hyperosmotic pressure enhances immunoglobulin transcription rates and secretion rates of KR12H-2 transfectoma. Biotechnol. Bioeng. 2000, 68, 260–268. [Google Scholar]
- Fujita, N.; Tamura, A.; Higashidani, A.; Tonozuka, T.; Freeze, H.H.; Nishikawa, A. The relative contribution of mannose salvage pathways to glycosylation in PMI-deficient mouse embryonic fibroblast cells. FEBS J. 2008, 275, 788–798. [Google Scholar]
- Kontoravdi, C.; Pistikopoulos, E.N.; Mantalaris, A. Systematic development of predictive mathematical models for animal cell cultures. Comput. Chem. Eng. 2010, 34, 1192–1198. [Google Scholar]
- Sobol, I.M. Global sensitivity indices for nonlinear mathematical models and their Monte Carlo estimates. Math. Comput. Simulat. 2001, 55, 271–280. [Google Scholar]
- Kontoravdi, C.; Asprey, S.P.; Pistikopoulos, E.N.; Mantalaris, A. Application of global sensitivity analysis to determine goals for design of experiments: An example study on antibody-producing cell cultures. Biotechnol. Progr. 2005, 21, 1128–1135. [Google Scholar]
- Kiparissides, A.; Kucherenko, S.S.; Mantalaris, A.; Pistikopoulos, E.N. Global sensitivity analysis challenges in biological systems modeling. Ind. Eng. Chem. Res. 2009, 48, 7168–7180. [Google Scholar]
- Goudar, C.; Biener, R.; Boisart, C.; Heidemann, R.; Piret, J.; de Graaf, A.; Konstantinov, K. Metabolic flux analysis of CHO cells in perfusion culture by metabolite balancing and 2D [C-13 H-1] COSY NMR spectroscopy. Metab. Eng. 2010, 12, 138–149. [Google Scholar]
- Li, G.Y.; Wang, S.W.; Rabitz, H. Practical approaches to construct RS-HDMR component functions. J. Phys. Chem. A 2002, 106, 8721–8733. [Google Scholar]
- Li, G.Y.; Hu, J.S.; Wang, S.W.; Georgopoulos, P.G.; Schoendorf, J.; Rabitz, H. Random sampling-high dimensional model representation (RS-HDMR) and orthogonality of its different order component functions. J. Phys. Chem. A 2006, 110, 2474–2485. [Google Scholar]
- Li, G.Y.; Rabitz, H. Ratio control variate method for efficiently determining high-dimensional model representations. J. Comput. Chem. 2006, 27, 1112–1118. [Google Scholar]
- Feil, B.; Kucherenko, S.; Shah, N. Comparison of Monte Carlo and Quasi Monte Carlo Sampling Methods in High Dimensional Model Representation. Proceedings of the Simul: 2009 First International Conference on Advances in System Simulation, Porto, Portugal, 20–25 September 2009; pp. 12–17.
- Zuniga, M.M.; Kucherenko, S.; Shah, N. Metamodelling with independent and dependent inputs. Comput. Phys. Commun. 2013, 184, 1570–1580. [Google Scholar]
- Geiger, T.; Velic, A.; Macek, B.; Lundberg, E.; Kampf, C.; Nagaraj, N.; Uhlen, M.; Cox, J.; Mann, M. Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol. Cell. Proteomics 2013, 12, 1709–1722. [Google Scholar]
- Baycin-Hizal, D.; Tabb, D.L.; Chaerkady, R.; Chen, L.; Lewis, N.E.; Nagarajan, H.; Sarkaria, V.; Kumar, A.; Wolozny, D.; Colao, J.; et al. Proteomic analysis of Chinese hamster ovary cells. J. Proteome Res. 2012, 11, 5265–5276. [Google Scholar]
- Xu, X.; Nagarajan, H.; Lewis, N.E.; Pan, S.; Cai, Z.; Liu, X.; Chen, W.; Xie, M.; Wang, W.; Hammond, S.; et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 2011, 29, 735–741. [Google Scholar]
- Milla, M.E.; Clairmont, C.A.; Hirschberg, C.B. Reconstitution into proteoliposomes and partial purification of the Golgi apparatus membrane UDP-galactose UDP-xylose and UDP-glucuronic acid transport activities. J. Biol. Chem. 1992, 267, 103–107. [Google Scholar]
- Dietmair, S.; Timmins, N.E.; Gray, P.P.; Nielsen, L.K.; Kromer, J.O. Towards quantitative metabolomics of mammalian cells: Development of a metabolite extraction protocol. Anal. Biochem. 2010, 404, 155–164. [Google Scholar]
- Del Val, I.J.; Kyriakopoulos, S.; Polizzi, K.M.; Kontoravdi, C. An optimized method for extraction and quantification of nucleotides and nucleotide sugars from mammalian cells. Anal. Biochem. 2013, 443, 172–180. [Google Scholar]
- Dell, A.; Reason, A.J.; Khoo, K.H.; Panico, M.; McDowell, R.A.; Morris, H.R. Mass spectrometry of carbohydrate-containing biopolymers. Methods Enzymol. 1994, 230, 108–132. [Google Scholar]
- Jang-Lee, J.; North, S.J.; Sutton-Smith, M.; Goldberg, D.; Panico, M.; Morris, H.; Haslam, S.; Dell, A. Glycomic profiling of cells and tissues by mass spectrometry: Fingerprinting and sequencing methodologies. Methods Enzymol. 2006, 415, 59–86. [Google Scholar]
- Damerell, D.; Ceroni, A.; Maass, K.; Ranzinger, R.; Dell, A.; Haslam, S.M. The GlycanBuilder and GlycoWorkbench glycoinformatics tools: Updates and new developments. Biol. Chem. 2012, 393, 1357–1362. [Google Scholar]
- Ceroni, A.; Maass, K.; Geyer, H.; Geyer, R.; Dell, A.; Haslam, S.M. GlycoWorkbench: A tool for the computer-assisted annotation of mass spectra of glycans. J. Proteome Res. 2008, 7, 1650–1659. [Google Scholar]
- Williamson, D.H.; Brosnan, J.T. Methods of Enzymatic Analysis; Bergmeyer, H.U., Gawehn, K., Eds.; Verlag Chemie: New York, NY, USA; London, UK, 1974; Volume 4. [Google Scholar]







| Glycan type | GlcNAc | GalNAc | Man | Gal | Neu5Ac | Fuc |
|---|---|---|---|---|---|---|
| N-linked glycan | 2.896 | 0 | 5.813 | 0.759 | 0.516 | 0.332 |
| O-linked glycan | 0.156 | 1 | 0 | 1.156 | 1.543 | 0 |
| Glycan average (NNSD,glyc) | 1.579 | 0.481 | 3.018 | 0.950 | 1.010 | 0.173 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jedrzejewski, P.M.; Del Val, I.J.; Constantinou, A.; Dell, A.; Haslam, S.M.; Polizzi, K.M.; Kontoravdi, C. Towards Controlling the Glycoform: A Model Framework Linking Extracellular Metabolites to Antibody Glycosylation. Int. J. Mol. Sci. 2014, 15, 4492-4522. https://doi.org/10.3390/ijms15034492
Jedrzejewski PM, Del Val IJ, Constantinou A, Dell A, Haslam SM, Polizzi KM, Kontoravdi C. Towards Controlling the Glycoform: A Model Framework Linking Extracellular Metabolites to Antibody Glycosylation. International Journal of Molecular Sciences. 2014; 15(3):4492-4522. https://doi.org/10.3390/ijms15034492
Chicago/Turabian StyleJedrzejewski, Philip M., Ioscani Jimenez Del Val, Antony Constantinou, Anne Dell, Stuart M. Haslam, Karen M. Polizzi, and Cleo Kontoravdi. 2014. "Towards Controlling the Glycoform: A Model Framework Linking Extracellular Metabolites to Antibody Glycosylation" International Journal of Molecular Sciences 15, no. 3: 4492-4522. https://doi.org/10.3390/ijms15034492