Molecular Dynamics Simulation on the Conformational Transition of the Mad2 Protein from the Open to the Closed State

Abstract
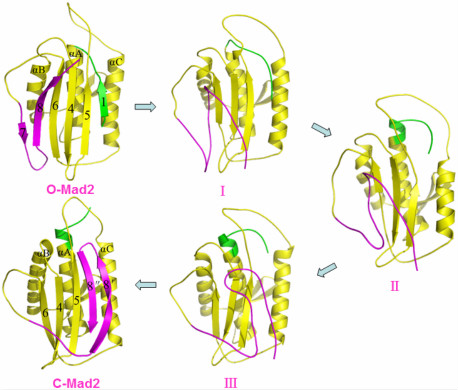
:
1. Introduction
2. Results
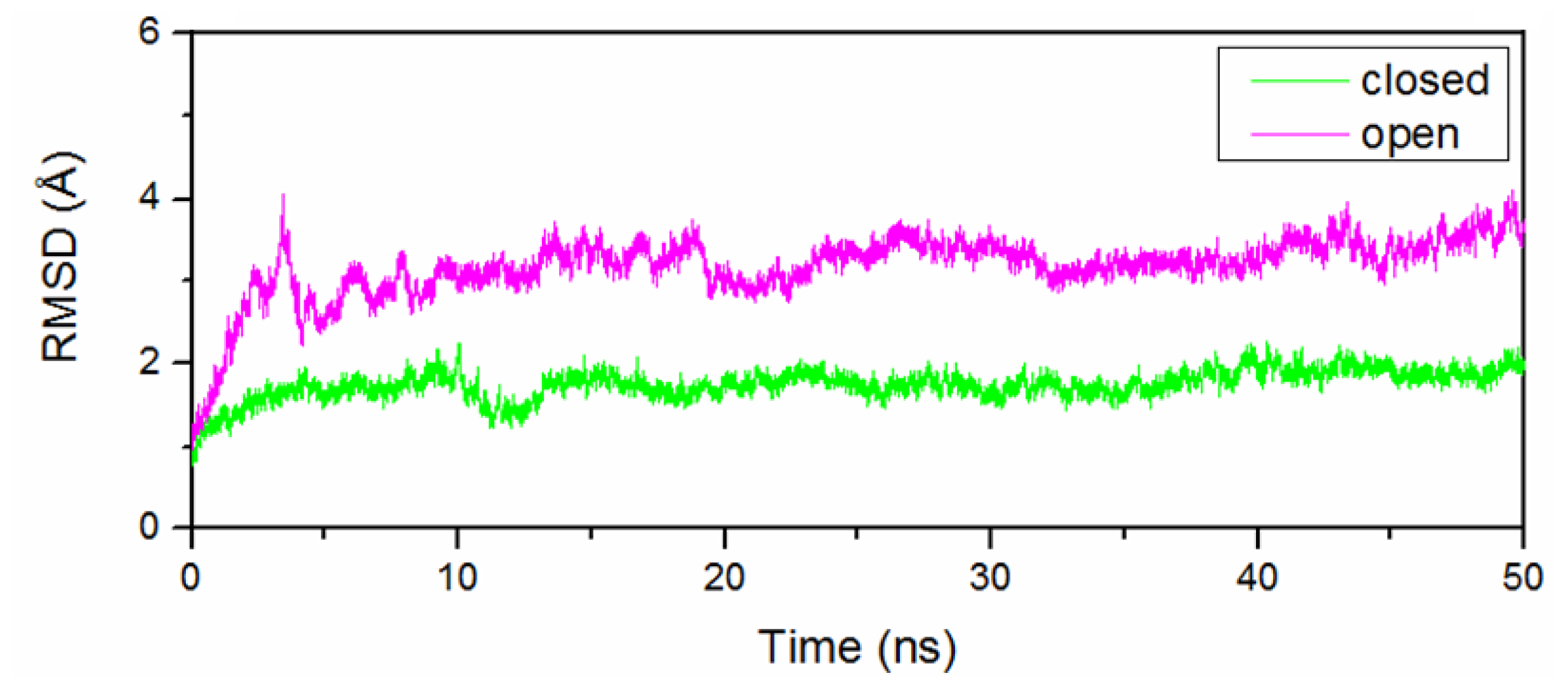
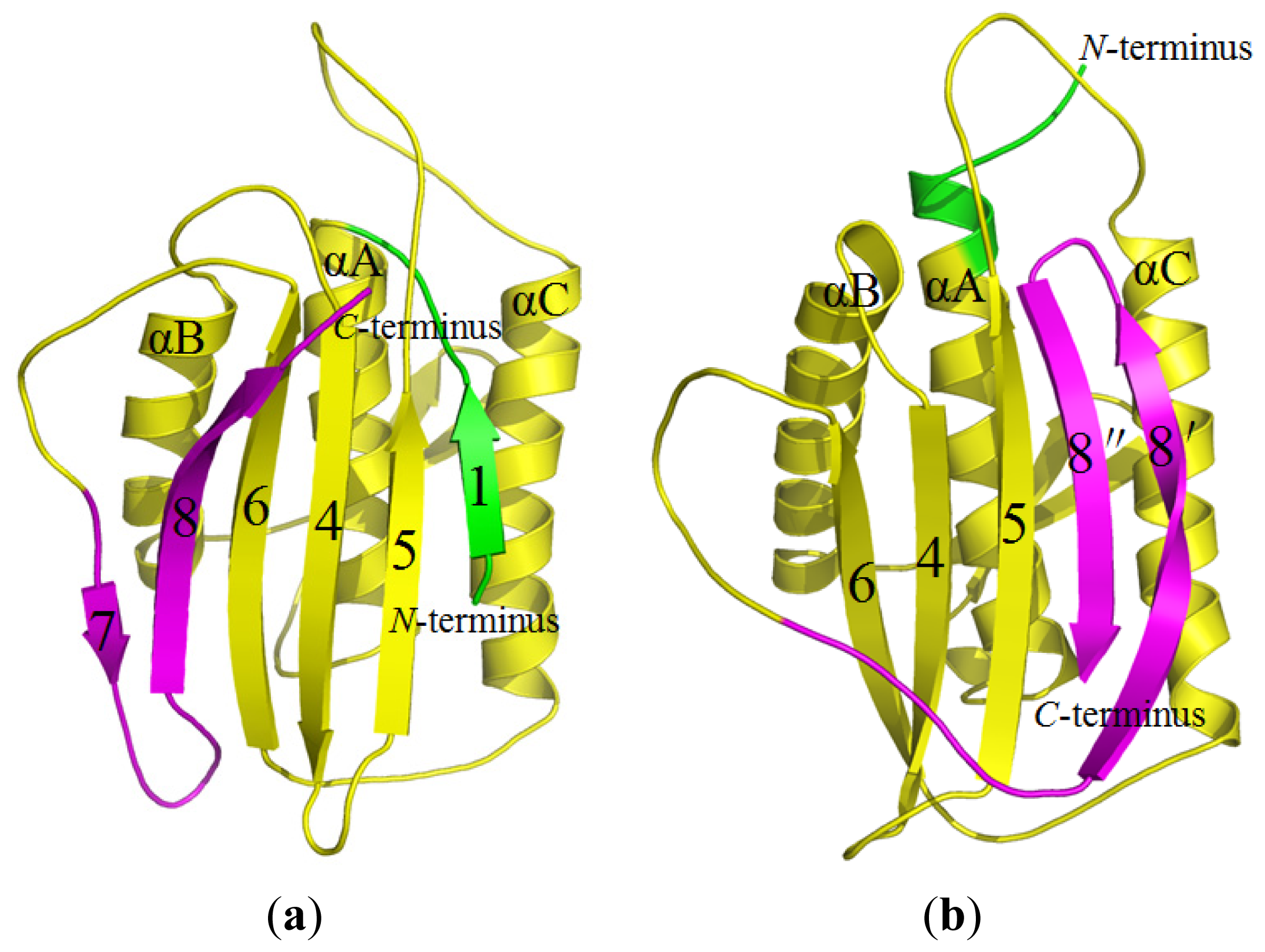
2.1. Stability of the O-Mad2 and C-Mad2 States
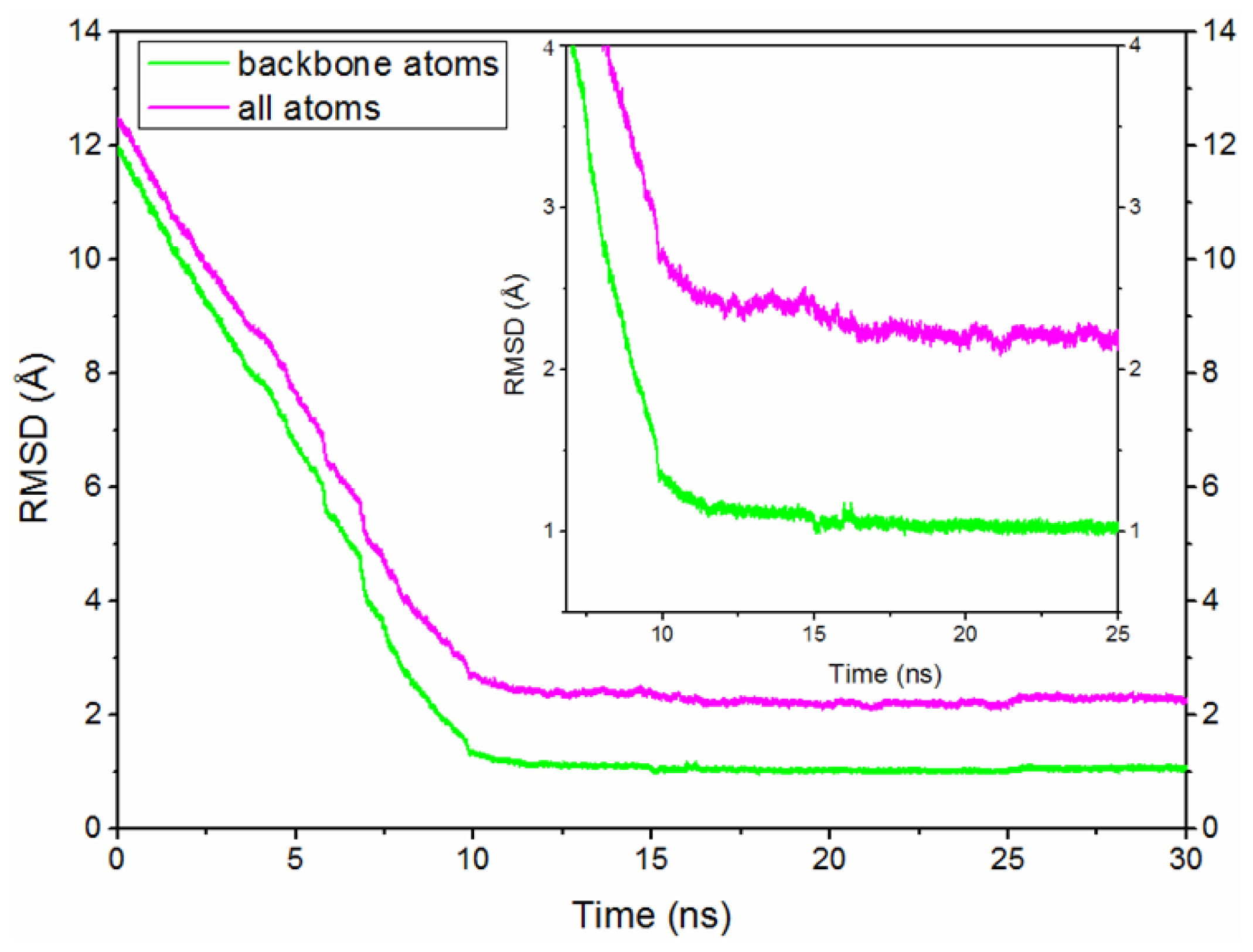
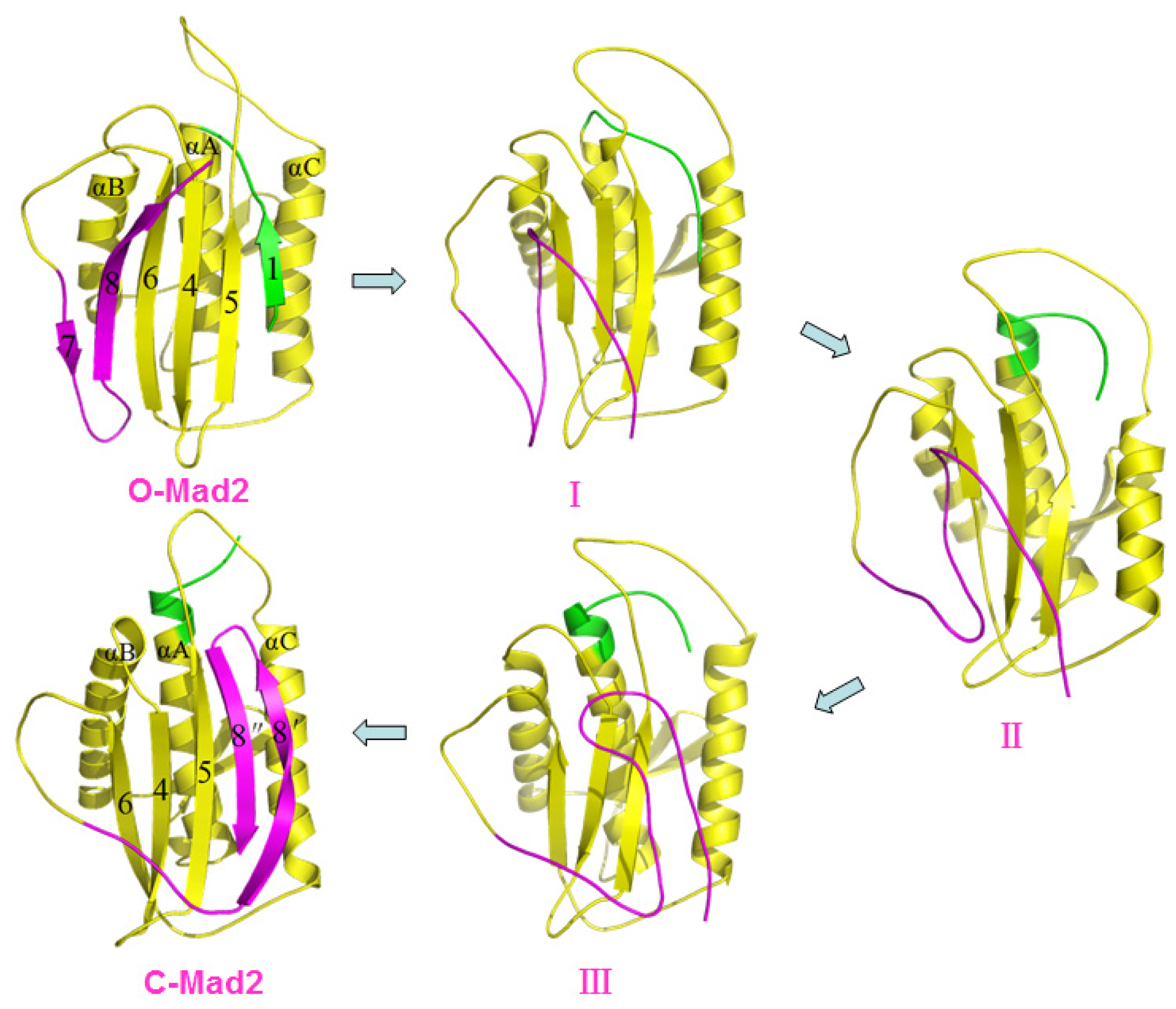
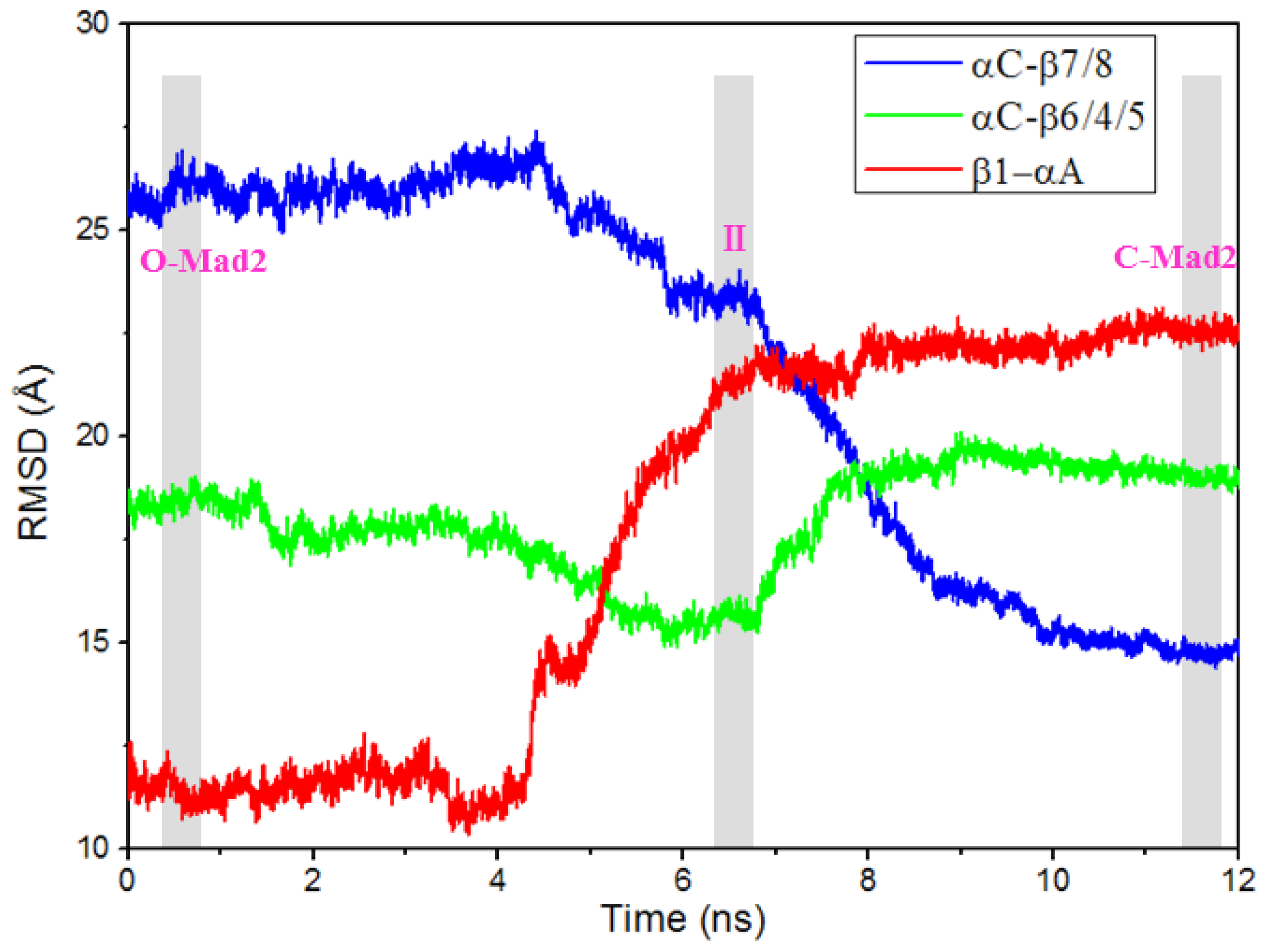
2.2. The Conformational Transition from O-Mad2 to C-Mad2
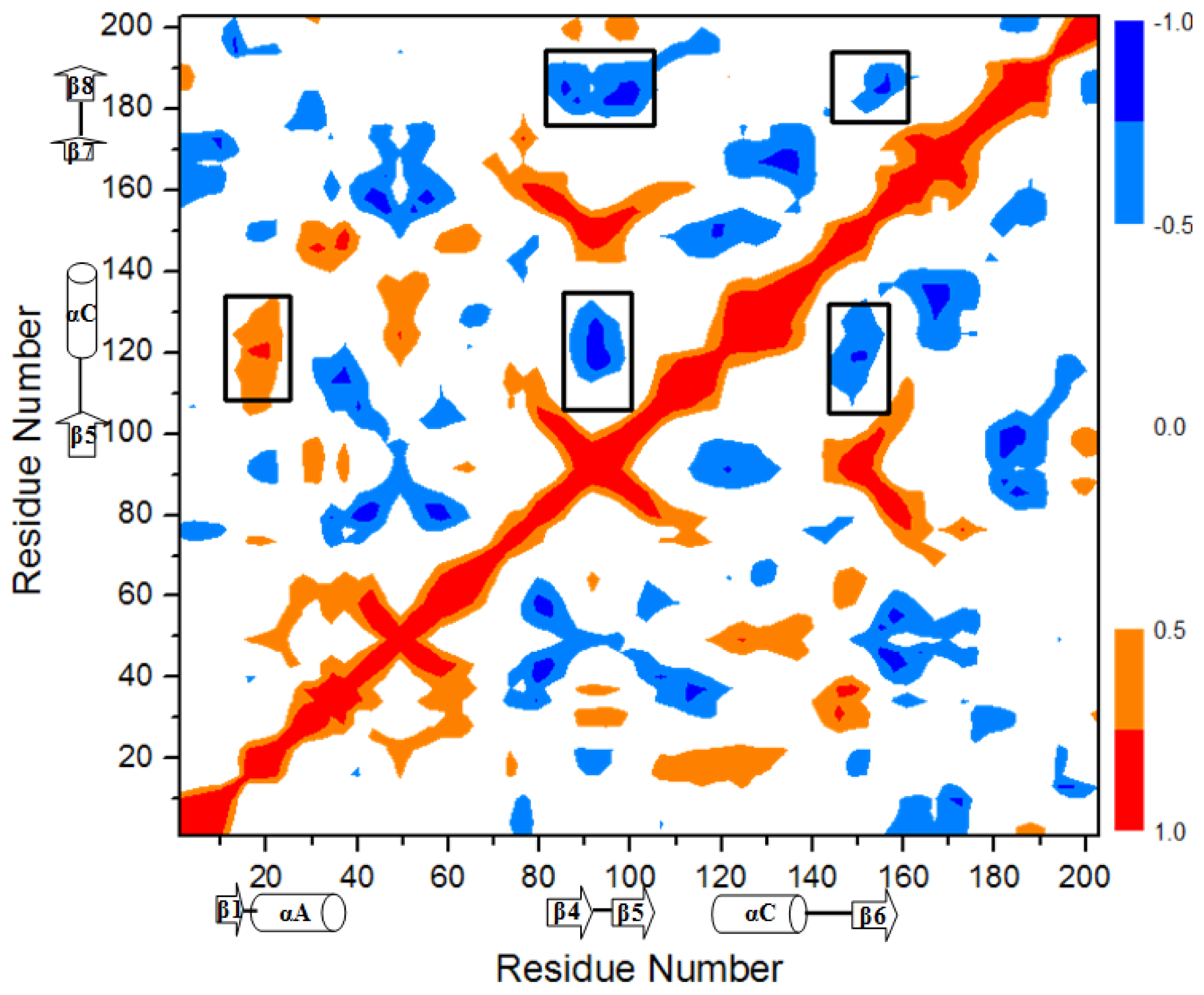
2.3. Correlation Analysis along the Transition Pathway from O-Mad2 to C-Mad2
3. Discussion
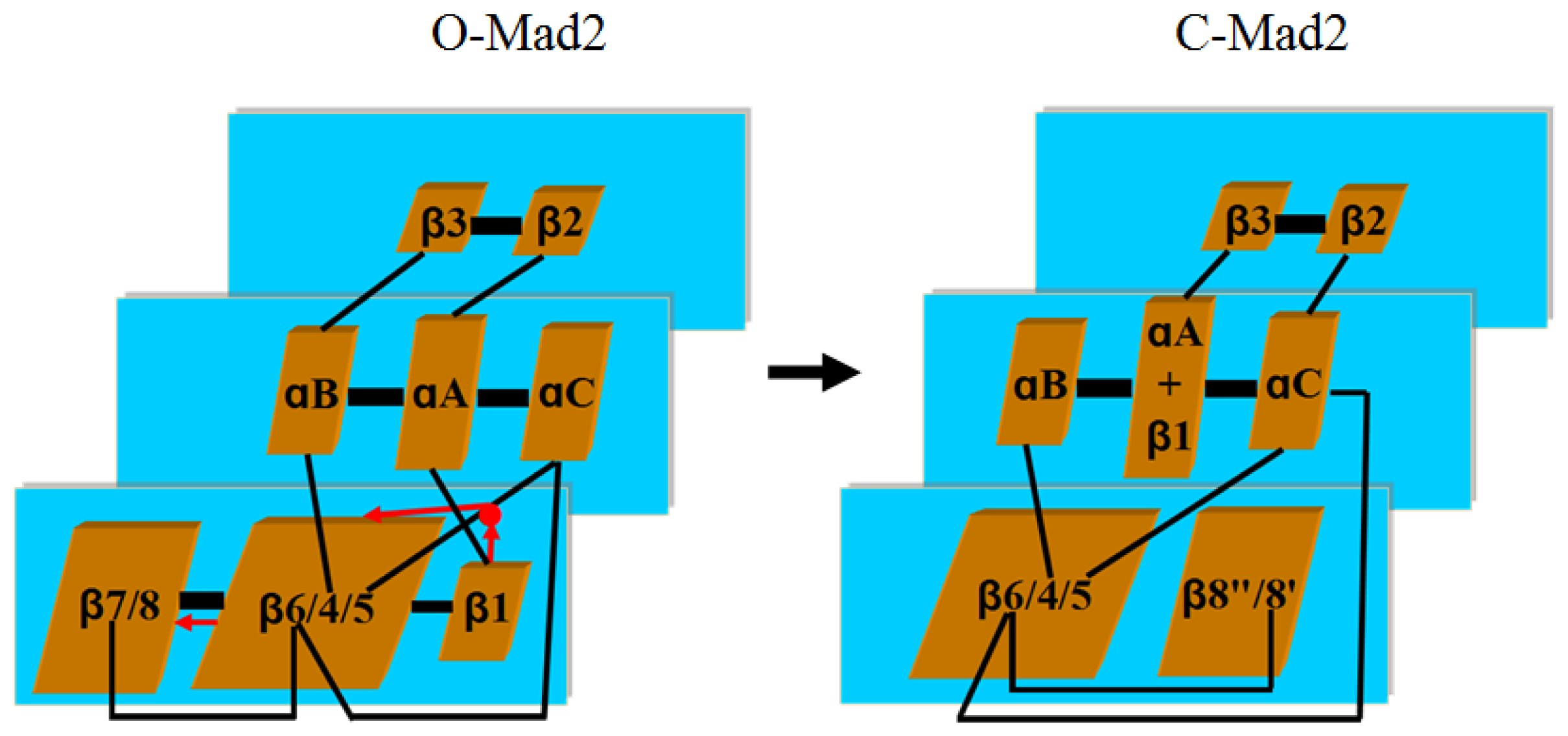
3.1. Conformational Characteristics along the Transition Pathway from O-Mad2 to C-Mad2
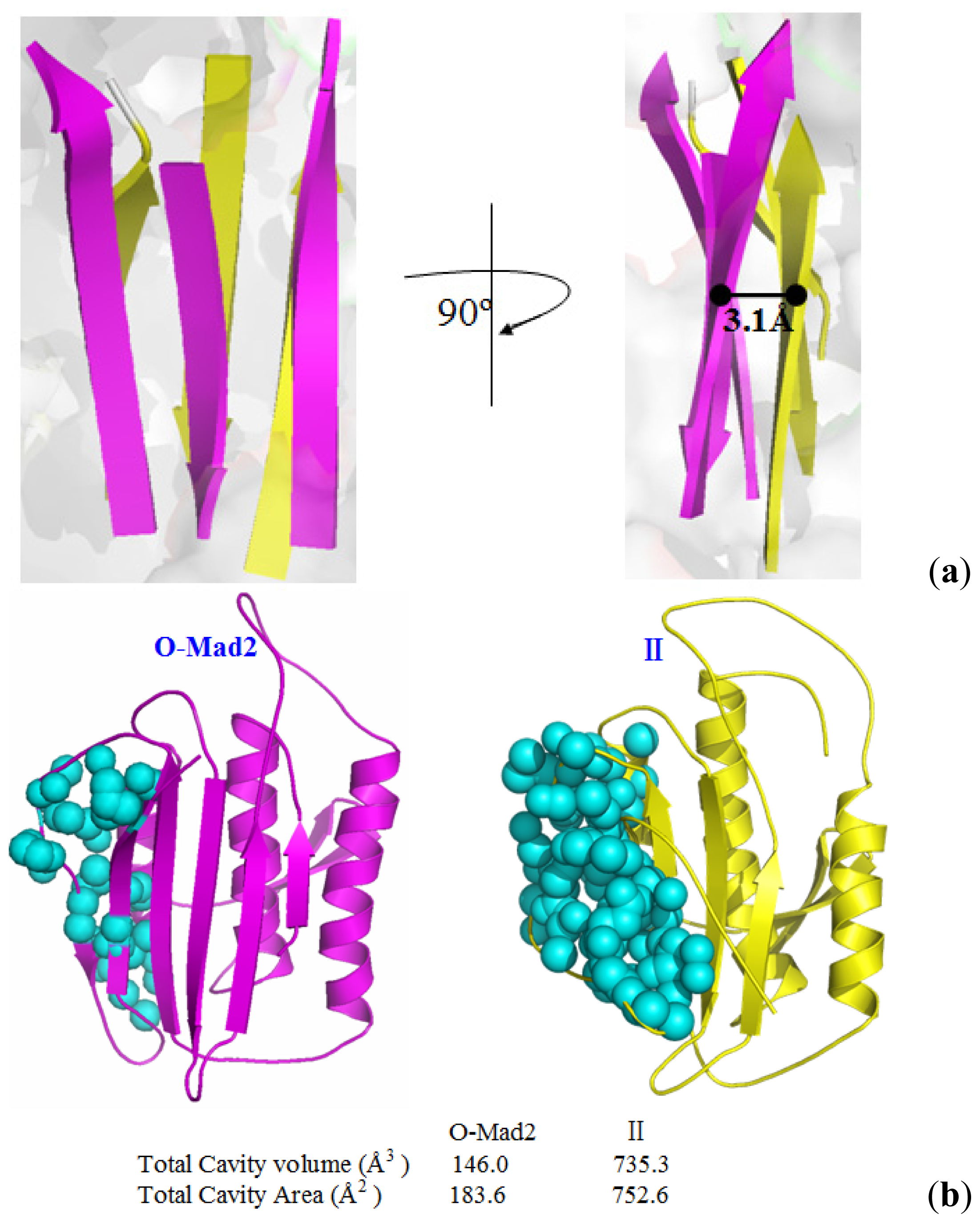
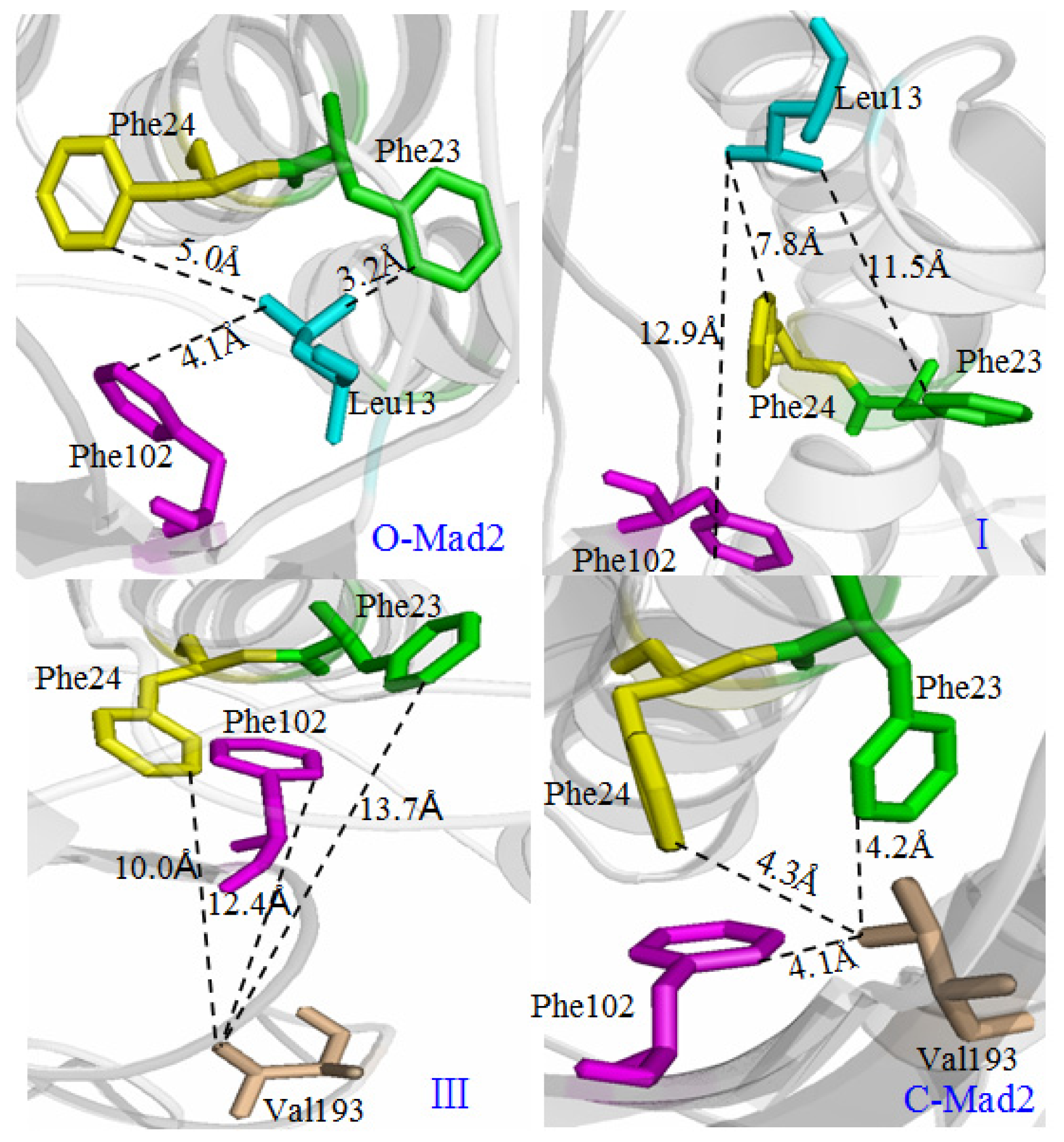
3.2. Hydrogen Bond and Hydrophobic Structure Analyses along the Transition Pathway from O-Mad2 to C-Mad2
3.3. Allosteric Network Analysis in the Conversion of O-Mad2 to C-Mad2
4. Models and Methods
4.1. Conventional Molecular Dynamics Simulation
4.2. Targeted Molecular Dynamics Simulation
4.3. Correlation of Atomic Motions
4.4. Surface Analyses
5. Conclusions
Supplementary Information
ijms-15-05553-s001.pdfAcknowledgments
Conflicts of Interest
- Author ContributionsConception and design: Y.W. and G.C.; Collection and assembly of data: C.L.; Data analysis and interpretation: C.L. and Y.Z.; and Manuscript writing: C.L. and Y.W.
References
- Luo, X.; Yu, H. Protein metamorphosis: The two-state behavior of Mad2. Structure 2008, 16, 1616–1625. [Google Scholar]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol 2007, 8, 379–393. [Google Scholar]
- Cleveland, D.W.; Mao, Y.; Sullivan, K.F. Centromeres and kinetochores: From epigenetics to mitotic checkpoint signaling. Cell 2003, 112, 407–422. [Google Scholar]
- Skladanowski, A.; Bozko, P.; Sabisz, M. DNA structure and integrity checkpoints during the cell cycle and their role in drug targeting and sensitivity of tumor cells to anticancer treatment. Chem. Rev 2009, 109, 2951–2973. [Google Scholar]
- Du, Y.; Yin, F.; Liu, C.; Hu, S.; Wang, J.; Xie, H.; Hong, L.; Fan, D. Depression of MAD2 inhibits apoptosis of gastric cancer cells by upregulating Bcl-2 and interfering mitochondrion pathway. Biochem. Biophys. Res. Commun 2006, 345, 1092–1098. [Google Scholar]
- Musacchio, A.; Hardwick, K.G. The spindle checkpoint: Structural insights into dynamic signalling. Nat. Rev. Mol. Cell Biol 2002, 3, 731–741. [Google Scholar]
- Peters, J.M. The anaphase promoting complex/cyclosome: A machine designed to destroy. Nat. Rev. Mol. Cell Biol 2006, 7, 644–656. [Google Scholar]
- Ditchfield, C.; Johnson, V.L.; Tighe, A.; Ellston, R.; Haworth, C.; Johnson, T.; Mortlock, A.; Keen, N.; Taylor, S.S. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol 2003, 161, 267–280. [Google Scholar]
- Shah, J.V.; Cleveland, D.W. Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell 2000, 103, 997–1000. [Google Scholar]
- Guardavaccaro, D.; Frescas, D.; Dorrello, N.V.; Peschiaroli, A.; Multani, A.S.; Cardozo, T.; Lasorella, A.; Iavarone, A.; Chang, S.; Hernando, E.; et al. Control of chromosome stability by the β-TrCP-REST-Mad2 axis. Nature 2008, 452, 365–369. [Google Scholar]
- Nezi, L.; Rancati, G.; de Antoni, A.; Pasqualato, S.; Piatti, S.; Musacchio, A. Accumulation of Mad2-Cdc20 complex during spindle checkpoint activation requires binding of open and closed conformers of Mad2 inSaccharomyces cerevisiae. J. Cell Biol 2006, 174, 39–51. [Google Scholar]
- Kulukian, A.; Han, J.S.; Cleveland, D.W. Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev. Cell 2009, 16, 105–117. [Google Scholar]
- Wickliffe, K.; Williamson, A.; Jin, L.; Rape, M. The multiple layers of ubiquitin-dependent cell cycle control. Chem. Rev 2009, 109, 1537–1548. [Google Scholar]
- Yang, M.; Li, B.; Liu, C.-J.; Tomchick, D.R.; Machius, M.; Rizo, J.; Yu, H.; Luo, X. Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol 2008, 6, e50. [Google Scholar]
- Takahashi, T.; Haruki, N.; Nomoto, S.; Masuda, A.; Saji, S.; Osada, H. Identification of frequent impairment of the mitotic checkpoint and molecular analysis of the mitotic checkpoint genes, hsMAD2 and p55CDC, in human lung cancers. Oncogene 1999, 18, 4295–4300. [Google Scholar]
- Bharadwaj, R.; Yu, H. The spindle checkpoint, aneuploidy, and cancer. Oncogene 2004, 23, 2016–2027. [Google Scholar]
- Wang, X.; Jin, D.-Y.; Ng, R.W.; Feng, H.; Wong, Y.C.; Cheung, A.L.; Tsao, S.W. Significance of MAD2 expression to mitotic checkpoint control in ovarian cancer cells. Cancer Res 2002, 62, 1662–1668. [Google Scholar]
- Kops, G.J.; Weaver, B.A.; Cleveland, D.W. On the road to cancer: Aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 2005, 5, 773–785. [Google Scholar]
- Murzin, A.G. Metamorphic proteins. Science 2008, 320, 1725–1726. [Google Scholar]
- Luo, X.; Tang, Z.; Xia, G.; Wassmann, K.; Matsumoto, T.; Rizo, J.; Yu, H. The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat. Struct. Mol. Biol 2004, 11, 338–345. [Google Scholar]
- Yang, M.; Li, B.; Tomchick, D.R.; Machius, M.; Rizo, J.; Yu, H.; Luo, X. p31comet blocks Mad2 activation through structural mimicry. Cell 2007, 131, 744–755. [Google Scholar]
- Mapelli, M.; Massimiliano, L.; Santaguida, S.; Musacchio, A. The Mad2 conformational dimer: Structure and implications for the spindle assembly checkpoint. Cell 2007, 131, 730–743. [Google Scholar]
- Luo, X.; Fang, G.; Coldiron, M.; Lin, Y.; Yu, H.; Kirschner, M.W.; Wagner, G. Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat. Struct. Mol. Biol 2000, 7, 224–229. [Google Scholar]
- Luo, X.; Tang, Z.; Rizo, J.; Yu, H. The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol. Cell 2002, 9, 59–71. [Google Scholar]
- Sironi, L.; Mapelli, M.; Knapp, S.; de Antoni, A.; Jeang, K.-T.; Musacchio, A. Crystal structure of the tetrameric Mad1–Mad2 core complex: Implications of a “safety belt” binding mechanism for the spindle checkpoint. EMBO J 2002, 21, 2496–2506. [Google Scholar]
- Mapelli, M.; Musacchio, A. MAD contortions: Conformational dimerization boosts spindle checkpoint signaling. Curr. Opin. Struct. Biol 2007, 17, 716–725. [Google Scholar]
- Fava, L.L.; Kaulich, M.; Nigg, E.A.; Santamaria, A. Probing the in vivo function of Mad1: C-Mad2 in the spindle assembly checkpoint. EMBO J 2011, 30, 3322–3336. [Google Scholar]
- Simonetta, M.; Manzoni, R.; Mosca, R.; Mapelli, M.; Massimiliano, L. The influence of catalysis on Mad2 activation dynamics. PLoS Biol 2009, 7, e1000010. [Google Scholar]
- Vink, M.; Simonetta, M.; Transidico, P.; Ferrari, K.; Mapelli, M.; de Antoni, A.; Massimiliano, L.; Ciliberto, A.; Faretta, M.; Salmon, E.D.; et al. In vitro FRAP identifies the minimal requirements for Mad2 kinetochore dynamics. Curr. Biol 2006, 16, 755–766. [Google Scholar]
- De Antoni, A.; Pearson, C.G.; Cimini, D.; Canman, J.C.; Sala, V.; Nezi, L.; Mapelli, M.; Sironi, L.; Faretta, M.; Salmon, E.D.; et al. The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr. Biol 2005, 15, 214–225. [Google Scholar]
- Lad, L.; Lichtsteiner, S.; Hartman, J.J.; Wood, K.W.; Sakowicz, R. Kinetic analysis of Mad2–Cdc20 formation: Conformational changes in Mad2 are catalyzed by a C-Mad2-ligand complex. Biochemistry 2009, 48, 9503–9515. [Google Scholar]
- Mapelli, M.; Filipp, F.V.; Rancati, G.; Massimiliano, L.; Nezi, L.; Stier, G.; Hagan, R.S.; Confalonieri, S.; Piatti, S.; Sattler, M. Determinants of conformational dimerization of Mad2 and its inhibition by p31comet. EMBO J 2006, 25, 1273–1284. [Google Scholar]
- Binkowski, T.A.; Naghibzadeh, S.; Liang, J. CASTp: Computed atlas of surface topography of proteins. Nucleic Acids Res 2003, 31, 3352–3355. [Google Scholar]
- Liang, J.; Woodward, C.; Edelsbrunner, H. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci 1998, 7, 1884–1897. [Google Scholar]
- Laskowski, R.A.; Luscombe, N.; Swindells, M.B.; Thornton, J.M. Protein clefts in molecular recognition and function. Protein Sci 1996, 5, 2438–2452. [Google Scholar]
- Case, D.; Darden, T.; Cheatham, T., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Merz, K.; Pearlman, D.; Crowley, M. AMBER 9; University of California: San Francisco, CA, USA, 2006. [Google Scholar]
- Lee, M.C.; Duan, Y. Distinguish protein decoys by using a scoring function based on a new AMBER force field, short molecular dynamics simulations, and the generalized born solvent model. Proteins 2004, 55, 620–634. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem 2003, 24, 1999–2012. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [Google Scholar]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem 1992, 13, 952–962. [Google Scholar]
- Schlitter, J.; Engels, M.; Krüger, P.; Jacoby, E.; Wollmer, A. Targeted molecular dynamics simulation of conformational change-application to the T↔R transition in insulin. Mol. Simul 1993, 10, 291–308. [Google Scholar]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph 1994, 12, 84–89. [Google Scholar]
- Sadiq, S.K.; de Fabritiis, G. Explicit solvent dynamics and energetics of HIV-1 protease flap opening and closing. Proteins 2010, 78, 2873–2885. [Google Scholar]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 2006, 34, W116–W118. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrogen bond | O-Mad2 | I | II | Hydrogen bond | III | C-Mad2 |
|---|---|---|---|---|---|---|
| (Ile11)N–H···O(Gln101) | 98.2 | 0.0 | – | (Arg99)N–H···O(Ala198) | 0.0 | 99.6 |
| (Gln101)N–H···O(Gln9) | 98.0 | 0.0 | – | (Arg99)O···H–N(Ala198) | 0.0 | 95.6 |
| (Asp103)N–H···O(Ile11) | 83.2 | 0.0 | – | (Gln101)N–H···O(Met196) | 0.0 | 100.0 |
| (Phe151)N–H···O(Ser185) | 98.0 | 0.0 | – | (Gln101)O···H–N(Met196) | 0.0 | 98.8 |
| (Leu153)N–H···O(Thr187) | 87.6 | 0.0 | – | (Asp103)N–H···O(Asn194) | 0.0 | 94.2 |
| (Ile155)N–H···O(Thr189) | 98.8 | 0.0 | – | (Asp103)O···H–N(Asn194) | 0.0 | 99.6 |
| (Thr157)N–H···O(His191) | 89.8 | 0.0 | – | (Glu105)N–H···O(Lys192) | 0.0 | 98.4 |
| (Thr188)N–H···O(Pro172) | 56.6 | 0.0 | – | (Glu105)O···H–N(Lys192) | 0.0 | 100.0 |
| (Thr189)N–H···O(Leu153) | 99.4 | 0.0 | – | (Glu179)N–H···O(Tyr199) | 0.0 | 99.4 |
| (His191)N–H···O(Ile155) | 99.4 | 0.0 | – | (Glu179)O···H–N(Tyr199) | 0.0 | 98.6 |
| (Thr187)N–H···O(Phe151) | 90.6 | 0.0 | – | (Val181)N–H···O(Val197) | 0.0 | 98.6 |
| (Phe186)N–H···O(Phe174) | 88.0 | 0.0 | – | (Val181)O···H–N(Val197) | 0.0 | 92.4 |
| (Phe186)O···H–N(Phe174) | 97.8 | 0.0 | – | (Leu183)N–H···O(Ser195) | 0.0 | 99.4 |
| (Ser16)N–H···O(Thr12) | – | 0.0 | 98.8 | (Phe186)N–H···O(Val193) | 0.0 | 99.4 |
| (Ala17)N–H···O(Leu13) | – | 0.0 | 85.7 | (Phe186)O···H–N(Val193) | 0.0 | 100.0 |
| (Glu18)N–H···O(Arg14) | – | 0.0 | 57.9 | (Thr188)N–H···O(His191) | 0.0 | 94.2 |
| (Ile19)N–H···O(Gly15) | – | 0.0 | 88.4 | (Lys200)N–H···O(Leu97) | 0.0 | 94.2 |
| (Val20)N–H···O(Ser16) | – | 0.0 | 99.2 | |||
| (Ala21)N–H···O(Ala17) | – | 0.0 | 54.5 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, C.; Zhu, Y.; Wang, Y.; Chen, G. Molecular Dynamics Simulation on the Conformational Transition of the Mad2 Protein from the Open to the Closed State. Int. J. Mol. Sci. 2014, 15, 5553-5569. https://doi.org/10.3390/ijms15045553
Li C, Zhu Y, Wang Y, Chen G. Molecular Dynamics Simulation on the Conformational Transition of the Mad2 Protein from the Open to the Closed State. International Journal of Molecular Sciences. 2014; 15(4):5553-5569. https://doi.org/10.3390/ijms15045553
Chicago/Turabian StyleLi, Chaoqun, Yanyan Zhu, Yan Wang, and Guangju Chen. 2014. "Molecular Dynamics Simulation on the Conformational Transition of the Mad2 Protein from the Open to the Closed State" International Journal of Molecular Sciences 15, no. 4: 5553-5569. https://doi.org/10.3390/ijms15045553