Frataxin Silencing Inactivates Mitochondrial Complex I in NSC34 Motoneuronal Cells and Alters Glutathione Homeostasis

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
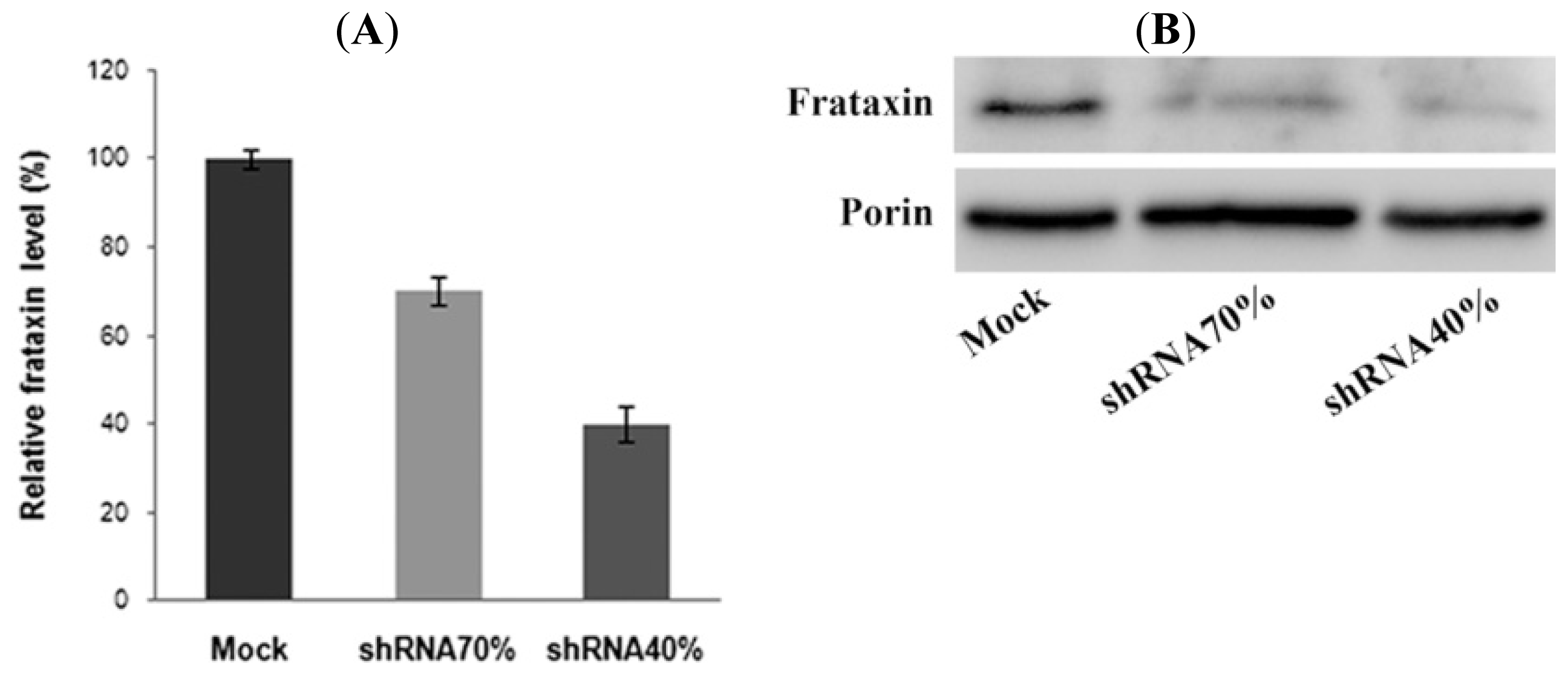
2.1. Frataxin Silencing in NSC34 Motor Neurons
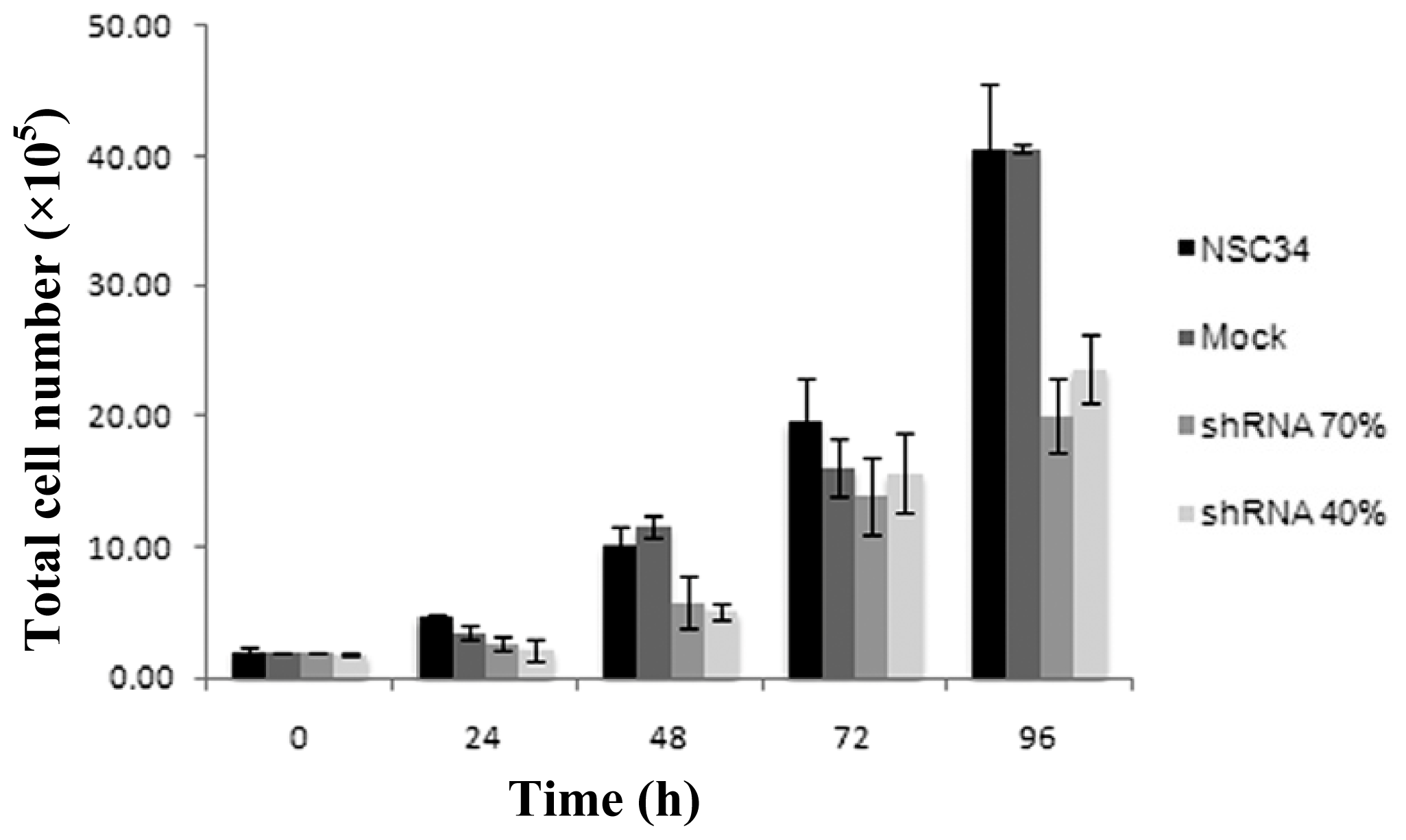
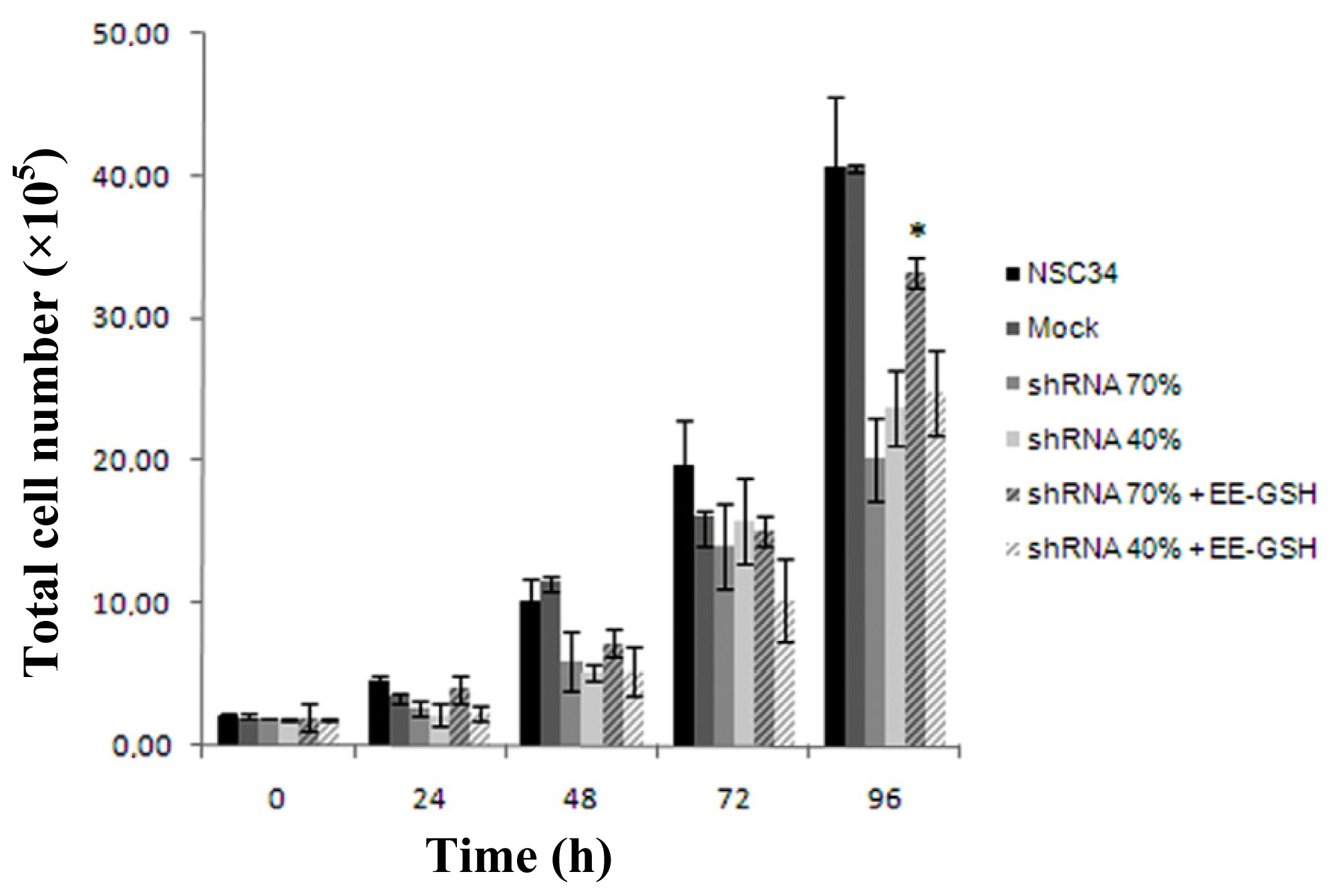
2.2. Frataxin-Silenced NSC34 Cells Show a Decrease of Cell Proliferation Rate
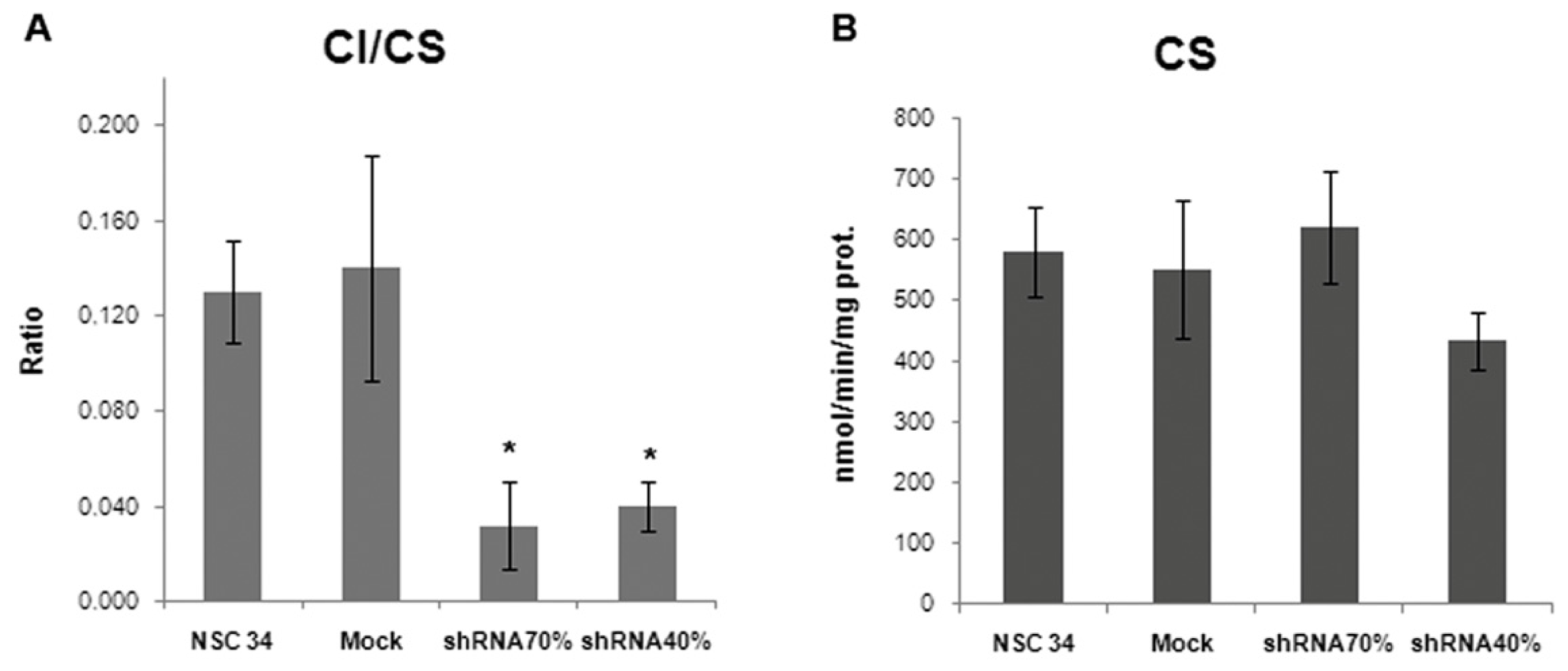
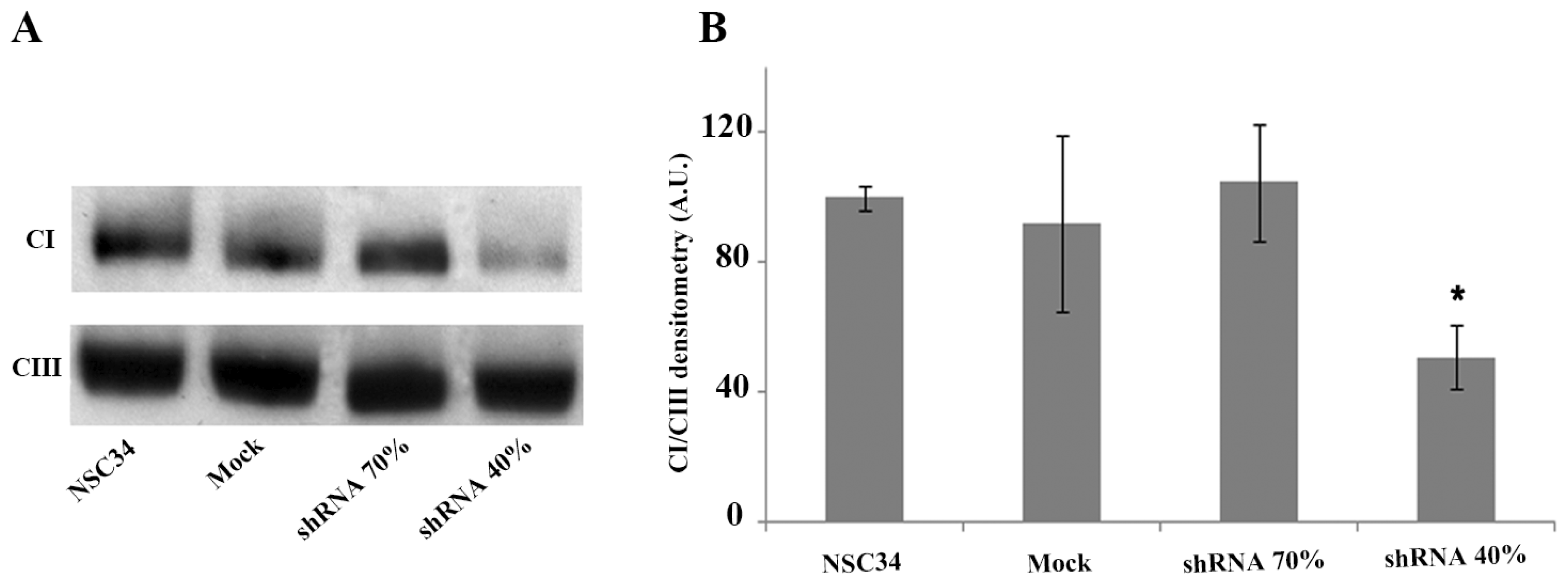
2.3. Frataxin Deficiency Specifically Affects the Mitochondrial Complex I
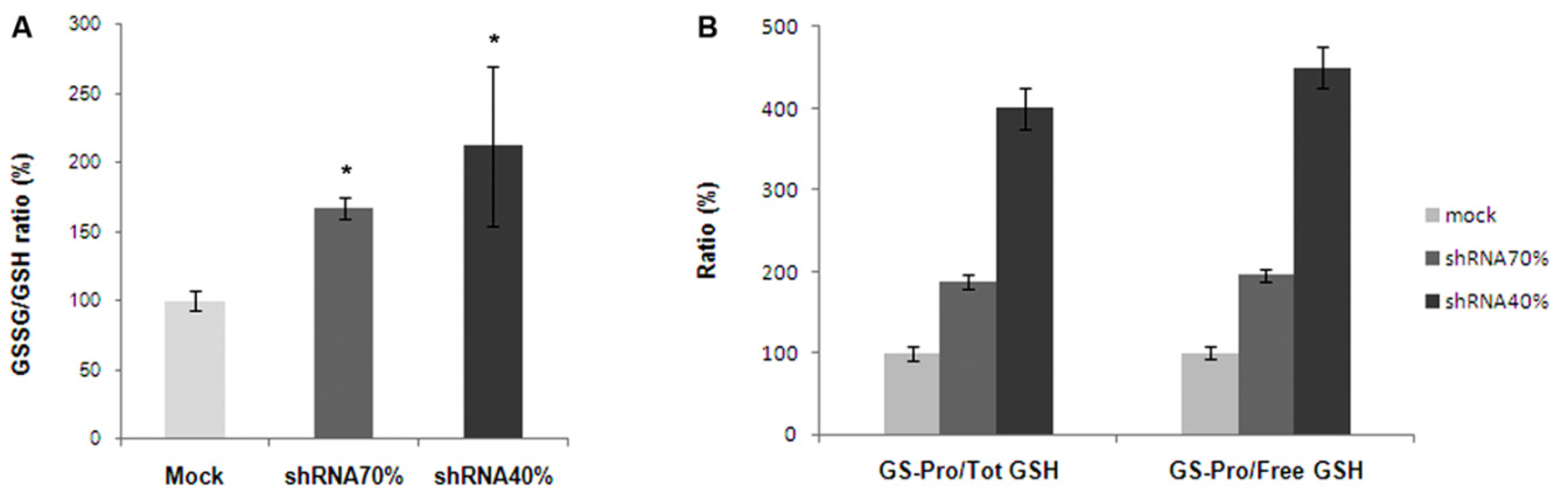
2.4. Frataxin Deficiency Alters the Glutathione Homeostasis in NSC34 Cells
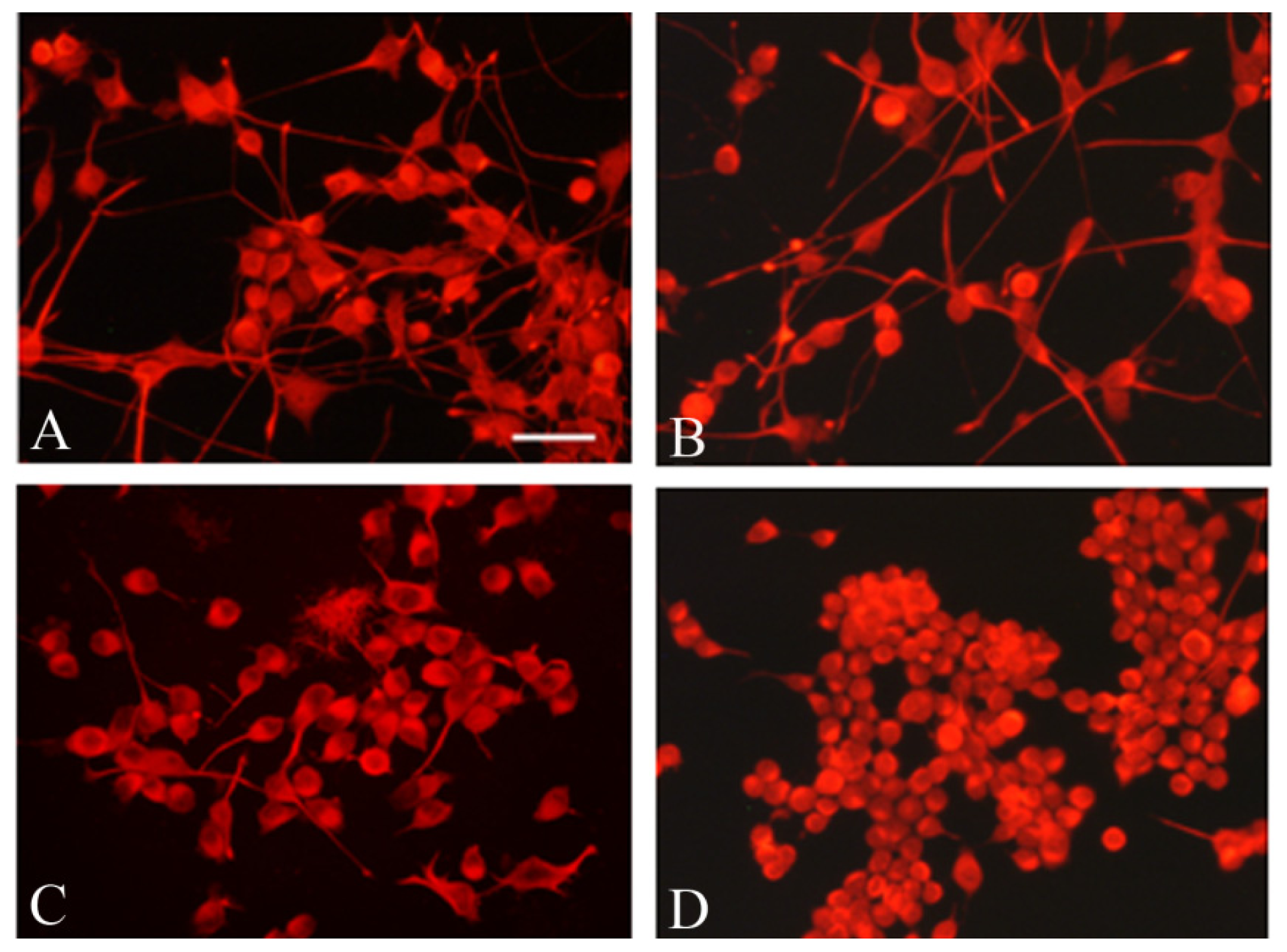
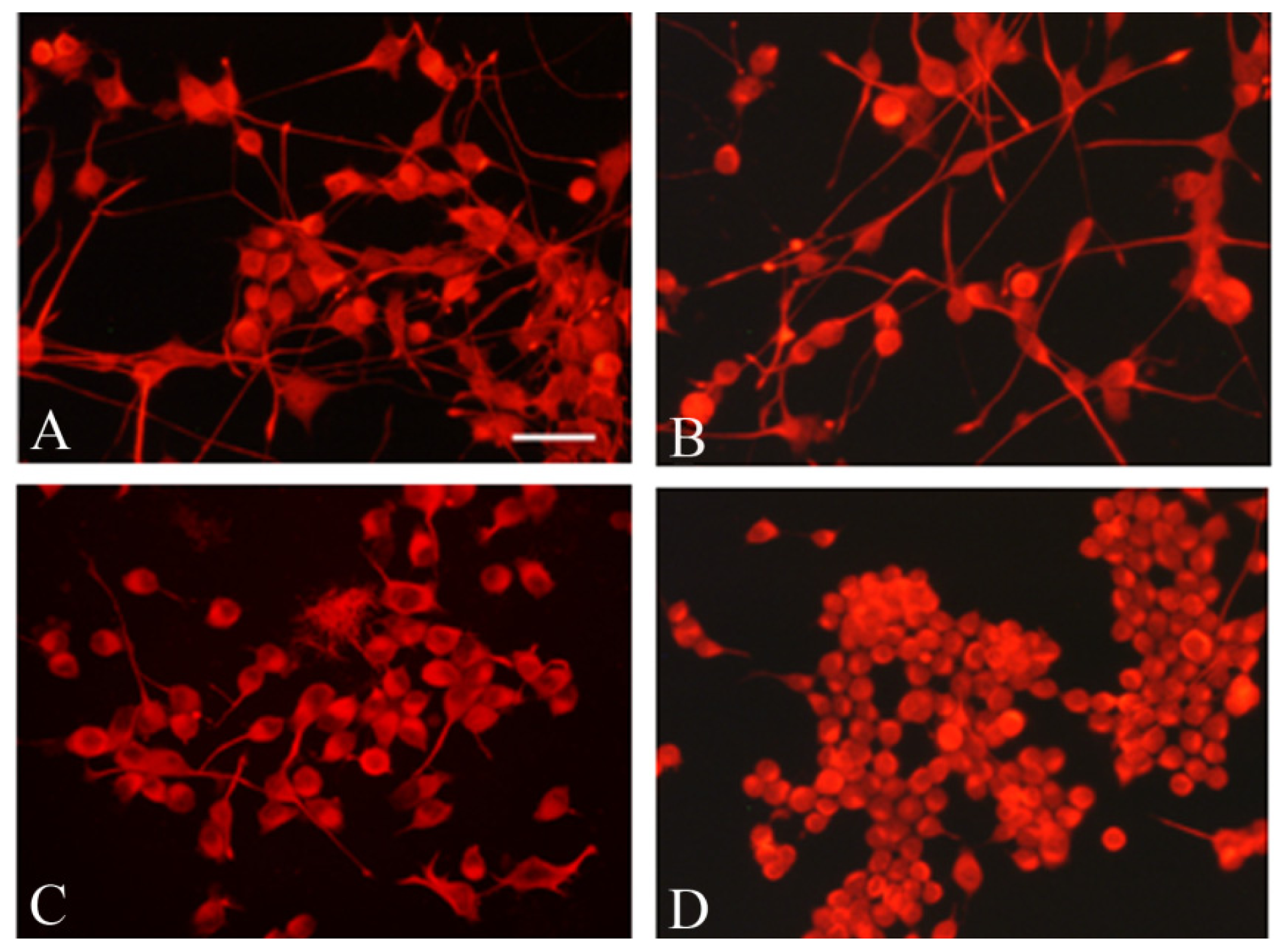
2.5. Frataxin Silencing Affects the Neuronal Morphology of NSC34 Cells
2.6. The in Vitro GSH-Treatment Restores the Proliferation Rate of Silenced NSC34 Cells
2.7. Discussion
3. Experimental Section
3.1. Double-Stranded RNA Design
3.2. Cell Culture
3.3. Stable shRNA Cell Lines Generation
3.4. Real-Time Quantitative PCR
3.5. Western Blot Analysis of Frataxin Levels
3.6. Cell Proliferation Assay
3.7. Immunocytochemistry
3.8. Mitochondria Isolation
3.9. Determination of Mitochondrial Respiratory Chain Enzyme Activities
3.10. HPLC Determination of Various Forms of Glutathione
3.11. Blue Native Gel Electrophoresis (BNGE)
3.12. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| FRDA | Friedreich’s Ataxia |
| Nrf2 | NF-E2-related factor |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| CNS | central nervous system |
| BNGE | blue native gel electrophoresis |
- Author ContributionsF.P. conceived of and led the project, and wrote the manuscript with B.C. B.C. together with F.P. and E.P. designed the experiments. B.C. developed NSC34 frataxin-silenced model, and with E.P. performed neuronal differentiation, cell proliferation assays and immunocitochemistry. G.T. with E.P. carried out the mitochondrial respiratory enzyme assays. A.P. and S.P. realized the HPLC determinations of glutathione forms. L.T. performed the Real-time quantitative PCR. A.T. the blue native gel electrophoresis. M.S., R.C. and E.B. supervised and contributed to the draft of the manuscript.
References
- Martelli, A.; Wattenhofer-Donzé, M.; Schmucker, S.; Bouvet, S.; Reutenauer, L.; Puccio, H. Frataxin is essential for extramitochondrial Fe–S cluster proteins in mammalian tissues. Hum. Mol. Genet 2007, 16, 2651–2658. [Google Scholar]
- Richardson, D.R.; Huang, M.L.; Whitnall, M.; Becker, E.M.; Ponka, P.; Suryo Rahmanto, Y. The ins and outs of mitochondrial iron-loading: The metabolic defect in Friedreich’s ataxia. J. Mol. Med 2010, 88, 323–329. [Google Scholar]
- Santos, R.; Lefevre, S.; Sliwa, D.; Seguin, A.; Camadro, J.M.; Lesuisse, E. Friedreich ataxia: Molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid. Redox Signal 2010, 13, 651–690. [Google Scholar]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhofer-Donzé, M.; Reutenauer, L.; Puccio, H. Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS One 2011, 6, e16199. [Google Scholar]
- Shan, Y.; Cortopassi, G. HSC20 interacts with frataxin and is involved in iron-sulfur cluster biogenesis and iron homeostasis. Hum. Mol. Genet 2012, 21, 1457–1469. [Google Scholar]
- Jauslin, M.L.; Wirth, T.; Meier, T.; Schoumacher, F.A. Cellular model for Friedreich ataxia reveals small-molecule glutathione peroxidase mimetics as novel treatment strategy. Hum. Mol. Genet 2002, 11, 3055–3063. [Google Scholar]
- Sturm, B.; Bistrich, U.; Schranzhofer, M.; Sarsero, J.P.; Rauen, U.; Scheiber-Mojdehkar, B.; de Groot, H.; Ioannou, P.; Petrat, F. Friedreich’s ataxia, no changes in mitochondrial labile iron in human lymphoblasts and fibroblasts: A decrease in antioxidative capacity? J. Biol. Chem 2005, 280, 6701–6708. [Google Scholar]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet 1999, 8, 425–430. [Google Scholar]
- Haugen, A.C.; di Prospero, N.A.; Parker, J.S.; Fannin, R.D.; Chou, J.; Meyer, J.N.; Halweg, C.; Collins, J.B.; Durr, A.; Fischbeck, K.; et al. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: Cellular model of pathology. PLoS Genet 2010, 6, e1000812. [Google Scholar]
- Zanella, I.; Derosas, M.; Corrado, M.; Cocco, E.; Cavadini, P.; Biasiotto, G.; Poli, M.; Verardi, R.; Arosio, P. The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochim. Biophys. Acta 2008, 1782, 90–98. [Google Scholar]
- Santos, M.M.; Ohshima, K.; Pandolfo, M. Frataxin deficiency enhances apoptosis in cells differentiating into neuroectoderm. Hum. Mol. Genet 2001, 10, 1935–1944. [Google Scholar]
- Calmels, N.; Schmucker, S.; Wattenhofer-Donzé, M.; Martelli, A.; Vaucamps, N.; Reutenauer, L.; Messaddeq, N.; Bouton, C.; Koenig, M.; Puccio, H. The first cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia. PLoS One 2009, 4, e6379. [Google Scholar]
- Lu, C.; Schoenfeld, R.; Shan, Y.; Tsai, H.J.; Hammock, B.; Cortopassi, G. Frataxin deficiency induces Schwann cell inflammation and death. Biochim. Biophys. Acta 2009, 1792, 1052–1061. [Google Scholar]
- Napoli, E.; Morin, D.; Bernhardt, R.; Buckpitt, A.; Cortopassi, G. Hemin rescues adrenodoxin, heme a and cytochrome oxidase activity in frataxin-deficient oligodendroglioma cells. Biochim. Biophys. Acta 2007, 1772, 773–780. [Google Scholar]
- Palomo, G.M.; Cerrato, T.; Gargini, R.; Diaz-Nido, J. Silencing of frataxin gene expression triggers p53-dependent apoptosis in human neuron-like cells. Hum. Mol. Genet 2011, 20, 2807–2822. [Google Scholar]
- Soragni, E.; Herman, D.; Dent, S.Y.; Gottesfeld, J.M.; Wells, R.D.; Napierala, M. Long intronic GAA*TTC repeats induce epigenetic changes and reporter gene silencing in a molecular model of Friedreich ataxia. Nucleic Acids Res 2008, 36, 6056–6065. [Google Scholar]
- Ku, S.; Soragni, E.; Campau, E.; Thomas, E.A.; Altun, G.; Laurent, L.C.; Loring, J.F.; Napierala, M.; Gottesfeld, J.M. Friedreich’s ataxia induced pluripotent stem cells model intergenerational GAA*TTC triplet repeat instability. Cell Stem Cell 2010, 7, 631–637. [Google Scholar]
- Liu, J.; Verma, P.J.; Evans-Galea, M.V.; Delatycki, M.B.; Michalska, A.; Leung, J.; Crombie, D.; Sarsero, J.P.; Williamson, R.; Dottori, M.; et al. Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev 2011, 7, 703–713. [Google Scholar]
- Hick, A.; Wattenhofer-Donzé, M.; Chintawar, S.; Tropel, P.; Simard, J.P.; Vaucamps, N.; Gall, D.; Lambot, L.; André, C.; Reutenauer, L.; et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich's ataxia. Dis. Model. Mech 2013, 6, 608–621. [Google Scholar]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraftm, A.D.; Lee, J.M.; Li, J.; Johnson, J.A. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal 2009, 11, 497–508. [Google Scholar]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci 2013, 14, 7853–7865. [Google Scholar]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar]
- Armstrong, J.S.; Khdour, O.; Hecht, S.M. Does oxidative stress contribute to the pathology of Friedreich’s ataxia? A radical question. FASEB J 2010, 24, 2152–2163. [Google Scholar]
- Rotig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial Fe/S protein deficiency in Friedreich ataxia. Nat. Genet 1997, 17, 215–217. [Google Scholar]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit intramitochondrial cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by iron deposits. Nat. Genet 2001, 27, 181–186. [Google Scholar]
- Auchère, F.; Santos, R.; Planamente, S.; Lesuisse, E.; Camadro, J.M. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich’s ataxia. Hum. Mol. Genet 2008, 17, 2790–2802. [Google Scholar]
- Bulteau, A.L.; Dancis, A.; Gareil, M.; Montagne, J.J.; Camadro, J.M.; Lesuisse, E. Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic. Biol. Med 2007, 42, 1561–1570. [Google Scholar]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem 2003, 278, 42588–42595. [Google Scholar]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Investig 2001, 31, 1007–1011. [Google Scholar]
- Edmond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar]
- Schulz, J.B.; Dehmar, T.; Schols, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar]
- Schulz, J.B.; Boesch, S.; Bürk, K.; Dürr, A.; Giunti, P.; Mariotti, C.; Pousset, F.; Schöls, L.; Vankan, P.; Pandolfo, M. Diagnosis and treatment of Friedreich ataxia: A European perspective. Nat. Rev. Neurol 2009, 5, 222–234. [Google Scholar]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal 2013, 19, 1481–1493. [Google Scholar]
- Calabrese, V.; Lodi, R.; Tonon, C.; D’Agata, V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.M.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci 2005, 233, 145–162. [Google Scholar]
- Lodi, R.; Tonon, C.; Calabrese, V.; Schapira, A.H. Friedreich’s ataxia: From disease mechanisms to therapeutic interventions. Antioxid. Redox Signal 2006, 8, 438–443. [Google Scholar]
- Llorens, J.V.; Navarro, J.A.; Martínez-Sebastián, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.A.; Moltó, M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J 2007, 21, 333–344. [Google Scholar]
- Carletti, B.; Passarelli, C.; Sparaco, M.; Tozzi, G.; Pastore, A.; Bertini, E.; Piemonte, F. Effect of protein glutathionylation on neuronal cytoskeleton: A potential link to neurodegeneration. Neuroscience 2011, 192, 285–294. [Google Scholar]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet 2000, 9, 275–282. [Google Scholar]
- Heidari, M.M.; Houshmand, M.; Hosseinkhani, S.; Nafissi, S.; Khatami, M. Complex I and ATP content deficiency in lymphocytes from Friedreich’s ataxia. Can. J. Neurol. Sci 2009, 36, 26–31. [Google Scholar]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of Complex I increases mitochondrial superoxide formation. J. Biol. Chem 2003, 278, 19603–19610. [Google Scholar]
- Papa, S.; de Rasmo, D.; Scacco, S.; Signorile, A.; Technikova-Dobrova, Z.; Palmisano, G.; Sardanelli, A.M.; Papa, F.; Panelli, D.; Scaringi, R.; et al. Mammalian Complex I: A regulable and vulnerable pacemaker in mitochondrial respiratory function. Biochim. Biophys. Acta 2008, 1777, 719–728. [Google Scholar]
- Mieyal, J.J.; Chock, P.B. Posttranslational modification of cysteine in redox signaling and oxidative stress: Focus on S-glutathionylation. Antioxid. Redox Signal 2012, 16, 471–475. [Google Scholar]
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases-focus on S-glutathionylation. Antioxid. Redox Signal 2012, 16, 543–566. [Google Scholar]
- Pastore, A.; Piemonte, F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci 2012, 46, 279–292. [Google Scholar]
- Dalle-Donne, I.; Rossi, R.; Milzani, A.; di Simplicio, P.; Colombo, R. The actin cytoskeleton responses to oxidants: From small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic. Biol. Med 2001, 31, 1624–1632. [Google Scholar]
- Sparaco, M.; Gaeta, L.M.; Santorelli, F.M.; Passarelli, C.; Tozzi, G.; Bertini, E.; Simonati, A.; Scaravilli, F.; Taroni, F.; Duyckaerts, C.; et al. Friedreich’s ataxia: Oxidative stress and cytoskeletal abnormalities. J. Neurol. Sci 2009, 287, 111–118. [Google Scholar]
- Sparaco, M.; Gaeta, L.M.; Tozzi, G.; Bertini, E.; Pastore, A.; Simonati, A.; Santorelli, F.M.; Piemonte, F. Protein glutathionylation in human central nervous system: Potential role in redox regulation of neuronal defense against free radicals. J. Neurosci. Res 2006, 83, 256–263. [Google Scholar]
- Pianese, L.; Turano, M.; lo Casale, M.S.; de Biase, I.; Giacchetti, M.; Monticelli, A.; Criscuolo, C.; Filla, A.; Cocozza, S. Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1061–1063. [Google Scholar]
- Santos, M.M.; Miranda, C.J.; Levy, J.E.; Montross, L.K.; Cossée, M.; Sequeiros, J.; Andrews, N.; Koenig, M.; Pandolfo, M. Iron metabolism in mice with partial frataxin deficiency. Cerebellum 2003, 2, 146–153. [Google Scholar]
- Coppola, G.; Choi, S.H.; Santos, M.M.; Miranda, C.J.; Tentler, D.; Wexler, E.M.; Pandolfo, M.; Geschwind, D.H. Gene expression profiling in frataxin deficient mice: Microarray evidence for significant expression changes without detectable neurodegeneration. Neurobiol. Dis 2006, 22, 302–311. [Google Scholar]
- Coppola, G.; Marmolino, D.; Lu, D.; Wang, Q.; Cnop, M.; Rai, M.; Acquaviva, F.; Cocozza, S.; Pandolfo, M.; Geschwind, D.H. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARγ pathway as a therapeutic target in Friedreich’s ataxia. Hum. Mol. Genet 2009, 18, 2452–2461. [Google Scholar]
- Coppola, G.; Burnett, R.; Perlman, S.; Versano, R.; Gao, F.; Plasterer, H.; Rai, M.; Saccá, F.; Filla, A.; Lynch, D.R.; et al. A gene expression phenotype in lymphocytes from Friedreich ataxia patients. Ann. Neurol 2011, 70, 790–804. [Google Scholar]
- Silva, J.M.; Li, M.Z.; Chang, K.; Ge, W.; Golding, M.C.; Rickles, R.J.; Siolas, D.; Hu, G.; Paddison, P.J.; Schlabach, M.R.; et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat. Genet 2005, 37, 1281–1288. [Google Scholar]
- Passarelli, C.; Tozzi, G.; Pastore, A.; Bertini, E.; Piemonte, F. GSSG-mediated complex I defect in isolated cardiac mitochondria. Int. J. Mol. Med 2010, 26, 95–99. [Google Scholar]
- Pastore, A.; Piemonte, F.; Locatelli, M.; lo Russo, A.; Gaeta, L.M.; Tozzi, G.; Federici, G. Determination of blood total, reduced, and oxidized glutathione in pediatric subjects. Clin. Chem 2001, 47, 1467–1469. [Google Scholar]
- Nijtmans, L.G.; Henderson, N.S.; Holt, I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods 2002, 26, 327–334. [Google Scholar]
- Eigentler, A.; Boesch, S.; Schneider, R.; Dechant, G.; Nat, R. Induced pluripotent stem cells from friedreich ataxia patients fail to upregulate frataxin during in vitro differentiation to peripheral sensory neurons. Stem Cells Dev 2013, 22, 3271–3282. [Google Scholar]
- Sandi, C.; Sandi, M.; Jassal, H.; Ezzatizadeh, V.; Anjomani-Virmouni, S.; Al-Mahdawi, S.; Pook, M.A. Generation and characterisation of Friedreich ataxia YG8R mouse fibroblast and neural stem cell models. PLoS One 2014, 9, e89488. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | NSC34 | Mock | shRNA 70% | shRNA 40% |
|---|---|---|---|---|
| CI | 0.13 ± 0.02 | 0.16 ± 0.05 | 0.03 ± 0.02 | 0.04 ± 0.01 * |
| CII | 0.08 ± 0.01 | 0.07 ± 0.02 | 0.06 ± 0.02 | 0.08 ± 0.02 |
| CIII | 0.21 ± 0.09 | 0.23 ± 0.07 | 0.18 ± 0.03 | 0.12 ± 0.03 |
| CIV | 1.97 ± 0.32 | 2.11 ± 0.36 | 1.83 ± 0.31 | 1.20 ± 0.20 |
| CS | 579 ± 73.71 | 550 ± 114.30 | 620 ± 91.40 | 434 ± 47.32 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carletti, B.; Piermarini, E.; Tozzi, G.; Travaglini, L.; Torraco, A.; Pastore, A.; Sparaco, M.; Petrillo, S.; Carrozzo, R.; Bertini, E.; et al. Frataxin Silencing Inactivates Mitochondrial Complex I in NSC34 Motoneuronal Cells and Alters Glutathione Homeostasis. Int. J. Mol. Sci. 2014, 15, 5789-5806. https://doi.org/10.3390/ijms15045789
Carletti B, Piermarini E, Tozzi G, Travaglini L, Torraco A, Pastore A, Sparaco M, Petrillo S, Carrozzo R, Bertini E, et al. Frataxin Silencing Inactivates Mitochondrial Complex I in NSC34 Motoneuronal Cells and Alters Glutathione Homeostasis. International Journal of Molecular Sciences. 2014; 15(4):5789-5806. https://doi.org/10.3390/ijms15045789
Chicago/Turabian StyleCarletti, Barbara, Emanuela Piermarini, Giulia Tozzi, Lorena Travaglini, Alessandra Torraco, Anna Pastore, Marco Sparaco, Sara Petrillo, Rosalba Carrozzo, Enrico Bertini, and et al. 2014. "Frataxin Silencing Inactivates Mitochondrial Complex I in NSC34 Motoneuronal Cells and Alters Glutathione Homeostasis" International Journal of Molecular Sciences 15, no. 4: 5789-5806. https://doi.org/10.3390/ijms15045789