Vasostatin Inhibits VEGF-Induced Endothelial Cell Proliferation, Tube Formation and Induces Cell Apoptosis under Oxygen Deprivation

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
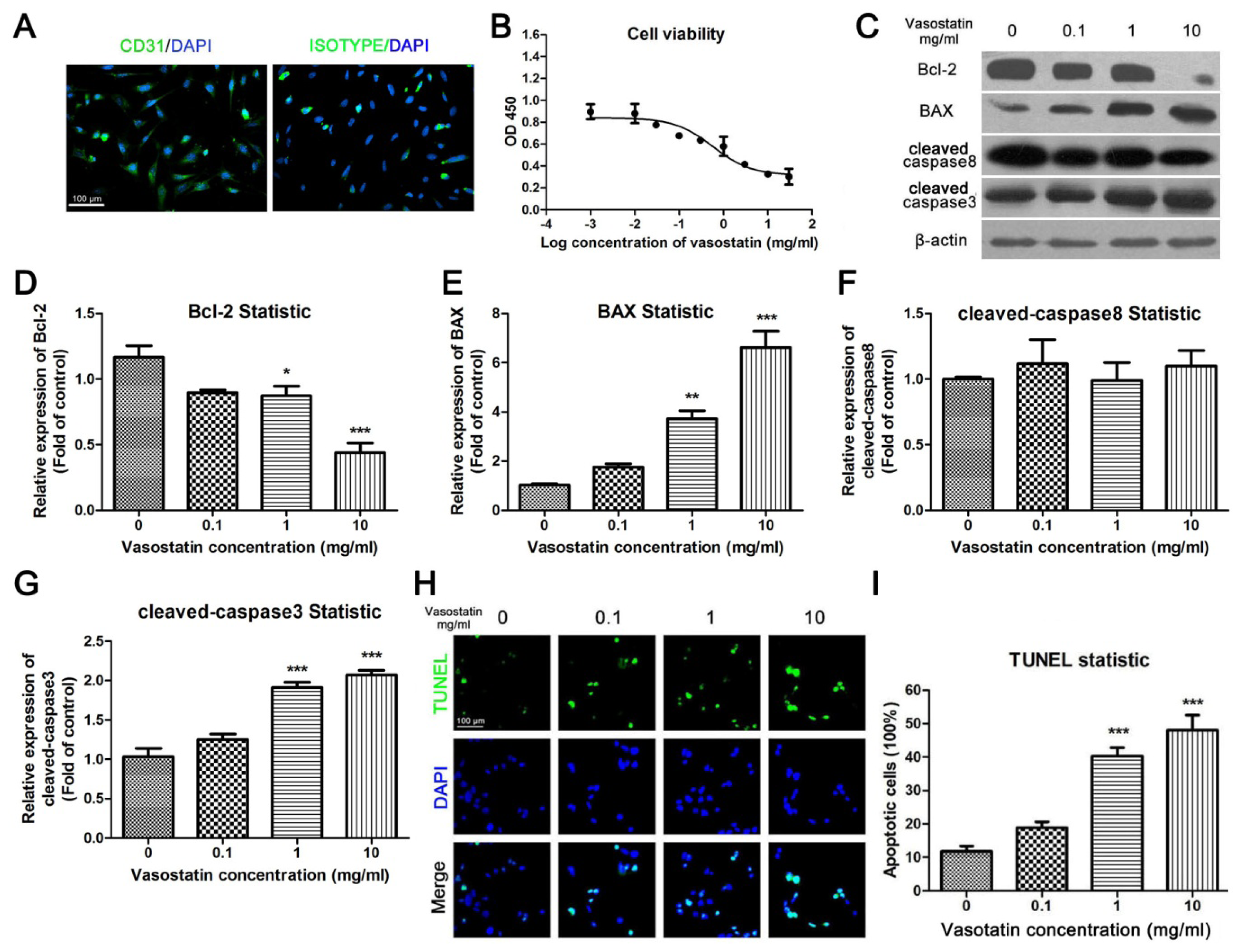
2.1. Vasostatin Inhibited Cell Viability Dose-Dependently and Induced Apoptosis of HUVEC under Oxygen-Deprivation
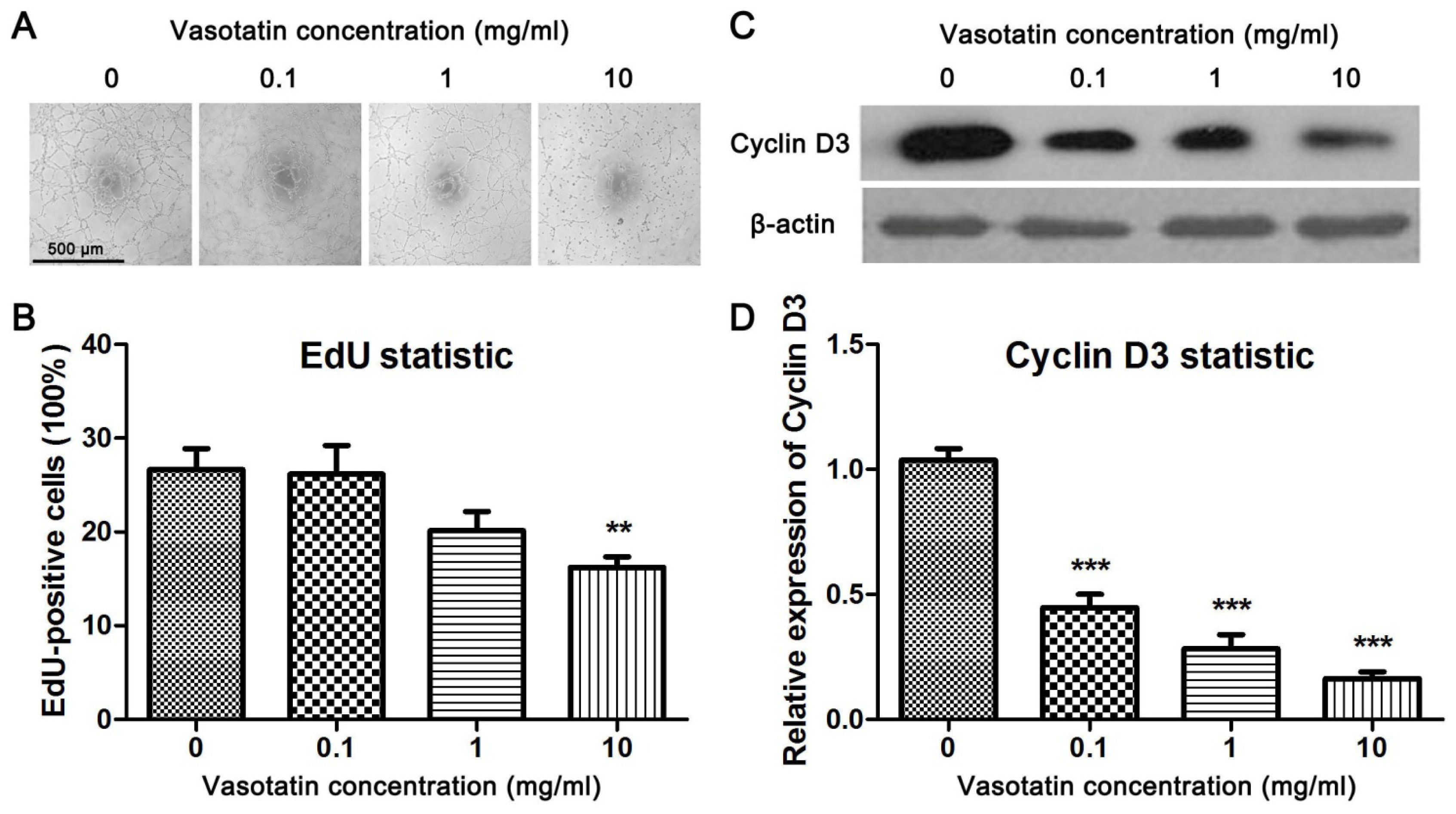
2.2. Vasostatin Inhibited VEGF-Induced Proliferation and Tube Formation of HUVEC
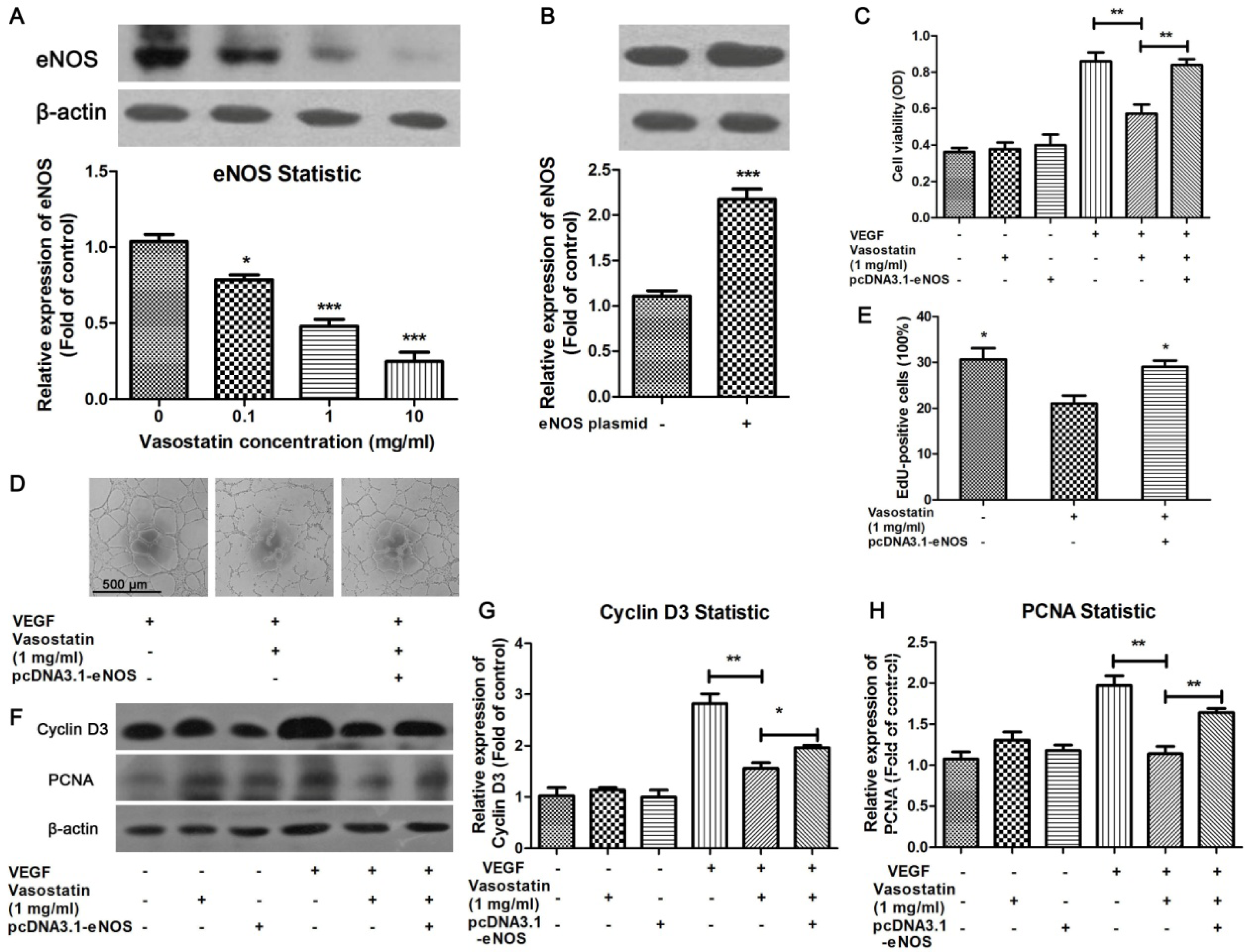
2.3. Overexpression of eNOS Reversed the Anti-Proliferation Effect of Vasostatin
2.4. Discussion
3. Experimental Section
3.1. Cell Isolation and Cultures
3.2. Cell Immunofluorescence and MTT Assay
3.3. 5-Ethynyl-2′-deoxyuridine (EdU) and TUNEL Staining
3.4. In Vitro Morphogenesis and Tube Formation Assay
3.5. Western Blot Analysis
3.6. Transient Transfection of pcDNA3.1-eNOS Plasmid
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsConceived and designed the experiments: Qun Shu. Performed the experiments and analyzed data: Qun Shu. Contribute reagents: Wenjiao Li, Haichuan Li. Wrote the paper: Qun Shu, Gang Sun.
References
- Folkman, J. Angiogenesis: Initiation and control. Ann. N. Y. Acad. Sci 1982, 401, 212–227. [Google Scholar]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar]
- Shojaei, F. Anti-angiogenesis therapy in cancer: Current challenges and future perspectives. Cancer Lett 2012, 320, 130–137. [Google Scholar]
- Pike, S.E.; Yao, L.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Teruya-Feldstein, J.; Wirth, P.; Gupta, G.; et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J. Exp. Med 1998, 188, 2349–2356. [Google Scholar]
- Yao, L.; Pike, S.E.; Setsuda, J.; Parekh, J.; Gupta, G.; Raffeld, M.; Jaffe, E.S.; Tosato, G. Effective targeting of tumor vasculature by the angiogenesis inhibitors vasostatin and interleukin-12. Blood 2000, 96, 1900–1905. [Google Scholar]
- Pike, S.E.; Yao, L.; Setsuda, J.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Atreya, C.D.; Teruya-Feldstein, J.; et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood 1999, 94, 2461–2468. [Google Scholar]
- Xiao, F.; Wei, Y.; Yang, L.; Zhao, X.; Tian, L.; Ding, Z.; Yuan, S.; Lou, Y.; Liu, F.; Wen, Y.; et al. A gene therapy for cancer based on the angiogenesis inhibitor, vasostatin. Gene Ther 2002, 9, 1207–1213. [Google Scholar]
- Tonra, J.R.; Hicklin, D.J. Targeting the vascular endothelial growth factor pathway in the treatment of human malignancy. Immunol. Investig 2007, 36, 3–23. [Google Scholar]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med 2012, 2. [Google Scholar] [CrossRef]
- Bogdanov, A.A., Jr.; Lin, C.P.; Kang, H.W. Optical imaging of the adoptive transfer of human endothelial cells in mice using anti-human CD31 monoclonal antibody. Pharm. Res 2007, 24, 1186–1192. [Google Scholar]
- Lamalice, L.; le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res 2007, 100, 782–794. [Google Scholar]
- Diermeier-Daucher, S.; Clarke, S.T.; Hill, D.; Vollmann-Zwerenz, A.; Bradford, J.A.; Brockhoff, G. Cell type specific applicability of 5-ethynyl-2′-deoxyuridine (EdU) for dynamic proliferation assessment in flow cytometry. Cytom. Part A 2009, 75, 535–546. [Google Scholar]
- Garcia-Cardena, G.; Fan, R.; Shah, V.; Sorrentino, R.; Cirino, G.; Papapetropoulos, A.; Sessa, W.C. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 1998, 392, 821–824. [Google Scholar]
- Gentile, C.; Muise-Helmericks, R.C.; Drake, C.J. VEGF-mediated phosphorylation of eNOS regulates angioblast and embryonic endothelial cell proliferation. Dev. Biol 2013, 373, 163–175. [Google Scholar]
- Patel, J.M. Calreticulin Regulation of Lung Endothelial NOS Activity; Landes Bioscience: Austin, TX, USA, 2000; pp. 205–219. [Google Scholar]
- Chiba, R.; Nakagawa, N.; Kurasawa, K.; Tanaka, Y.; Saito, Y.; Iwamoto, I. Ligation of CD31 (PECAM-1) on endothelial cells increases adhesive function of alphavbeta3 integrin and enhances beta1 integrin-mediated adhesion of eosinophils to endothelial cells. Blood 1999, 94, 1319–1329. [Google Scholar]
- Gallo, M.P.; Levi, R.; Ramella, R.; Brero, A.; Boero, O.; Tota, B.; Alloatti, G. Endothelium-derived nitric oxide mediates the antiadrenergic effect of human vasostatin-1 in rat ventricular myocardium. Am. J. Physiol. Heart Circ. Physiol 2007, 292, H2906–H2912. [Google Scholar]
- Wu, P.C.; Yang, L.C.; Kuo, H.K.; Huang, C.C.; Tsai, C.L.; Lin, P.R.; Shin, S.J.; Tai, M.H. Inhibition of corneal angiogenesis by local application of vasostatin. Mol. Vis 2005, 11, 28–35. [Google Scholar]
- Huegel, R.; Velasco, P.; de la Luz Sierra, M.; Christophers, E.; Schroder, J.M.; Schwarz, T.; Tosato, G.; Lange-Asschenfeldt, B. Novel anti-inflammatory properties of the angiogenesis inhibitor vasostatin. J. Investig. Dermatol 2007, 127, 65–74. [Google Scholar]
- Mans, S.; Banz, Y.; Mueller, B.U.; Pabst, T. The angiogenesis inhibitor vasostatin is regulated by neutrophil elastase-dependent cleavage of calreticulin in AML patients. Blood 2012, 120, 2690–2699. [Google Scholar]
- Halestrap, A.P.; Doran, E.; Gillespie, J.P.; O’Toole, A. Mitochondria and cell death. Biochem. Soc. Trans 2000, 28, 170–177. [Google Scholar]
- Mazars, A.; Geneste, O.; Hickman, J. The Bcl-2 family of proteins as drug targets. J. Soc. Biol 2005, 199, 253–265. [Google Scholar]
- Miller, D.K. The role of the Caspase family of cysteine proteases in apoptosis. Semin. Immunol 1997, 9, 35–49. [Google Scholar]
- MacLauchlan, S.; Yu, J.; Parrish, M.; Asoulin, T.A.; Schleicher, M.; Krady, M.M.; Zeng, J.; Huang, P.L.; Sessa, W.C.; Kyriakides, T.R. Endothelial nitric oxide synthase controls the expression of the angiogenesis inhibitor thrombospondin 2. Proc. Natl. Acad. Sci. USA 2011, 108, E1137–E1145. [Google Scholar]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig 1998, 101, 731–736. [Google Scholar]
- Yu, J.; deMuinck, E.D.; Zhuang, Z.; Drinane, M.; Kauser, K.; Rubanyi, G.M.; Qian, H.S.; Murata, T.; Escalante, B.; Sessa, W.C. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc. Natl. Acad. Sci. USA 2005, 102, 10999–11004. [Google Scholar]
- Murohara, T.; Witzenbichler, B.; Spyridopoulos, I.; Asahara, T.; Ding, B.; Sullivan, A.; Losordo, D.W.; Isner, J.M. Role of endothelial nitric oxide synthase in endothelial cell migration. Arterioscler. Thromb. Vasc. Biol 1999, 19, 1156–1161. [Google Scholar]
- Yao, L.; Pike, S.E.; Tosato, G. Laminin binding to the calreticulin fragment vasostatin regulates endothelial cell function. J. Leukoc. Biol 2002, 71, 47–53. [Google Scholar]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar]
- Dembinska-Kiec, A.; Polus, A.; Kiec-Wilk, B.; Grzybowska, J.; Mikolajczyk, M.; Hartwich, J.; Razny, U.; Szumilas, K.; Banas, A.; Bodzioch, M.; et al. Proangiogenic activity of beta-carotene is coupled with the activation of endothelial cell chemotaxis. Biochim. Biophys. Acta 2005, 1740, 222–239. [Google Scholar]
- Im, E.; Venkatakrishnan, A.; Kazlauskas, A. Cathepsin B regulates the intrinsic angiogenic threshold of endothelial cells. Mol. Biol. Cell 2005, 16, 3488–3500. [Google Scholar]
- Shen, J.; Qu, C.K. In vitro hematopoietic differentiation of murine embryonic stem cells. Methods Mol. Biol 2008, 430, 103–118. [Google Scholar]
- Kaur, S.; Kumar, T.R.; Uruno, A.; Sugawara, A.; Jayakumar, K.; Kartha, C.C. Genetic engineering with endothelial nitric oxide synthase improves functional properties of endothelial progenitor cells from patients with coronary artery disease: An in vitro study. Basic Res. Cardiol 2009, 104, 739–749. [Google Scholar]
- Qiao, T.; Liu, C.J.; Ran, F.; Han, L.; Zhang, L.; Li, L. Experimental study of recombinant eukaryotic expression vector of human eNOS in ECV304. Swiss Med. Wkly 2006, 136, 19–25. [Google Scholar]



© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shu, Q.; Li, W.; Li, H.; Sun, G. Vasostatin Inhibits VEGF-Induced Endothelial Cell Proliferation, Tube Formation and Induces Cell Apoptosis under Oxygen Deprivation. Int. J. Mol. Sci. 2014, 15, 6019-6030. https://doi.org/10.3390/ijms15046019
Shu Q, Li W, Li H, Sun G. Vasostatin Inhibits VEGF-Induced Endothelial Cell Proliferation, Tube Formation and Induces Cell Apoptosis under Oxygen Deprivation. International Journal of Molecular Sciences. 2014; 15(4):6019-6030. https://doi.org/10.3390/ijms15046019
Chicago/Turabian StyleShu, Qun, Wenjiao Li, Haichuan Li, and Gang Sun. 2014. "Vasostatin Inhibits VEGF-Induced Endothelial Cell Proliferation, Tube Formation and Induces Cell Apoptosis under Oxygen Deprivation" International Journal of Molecular Sciences 15, no. 4: 6019-6030. https://doi.org/10.3390/ijms15046019