Differential Signaling by Protease-Activated Receptors: Implications for Therapeutic Targeting

{kind=link}
Abstract
:1. Introduction
2. Exploiting Differences in PAR Activation Mechanisms for Therapeutic Benefit
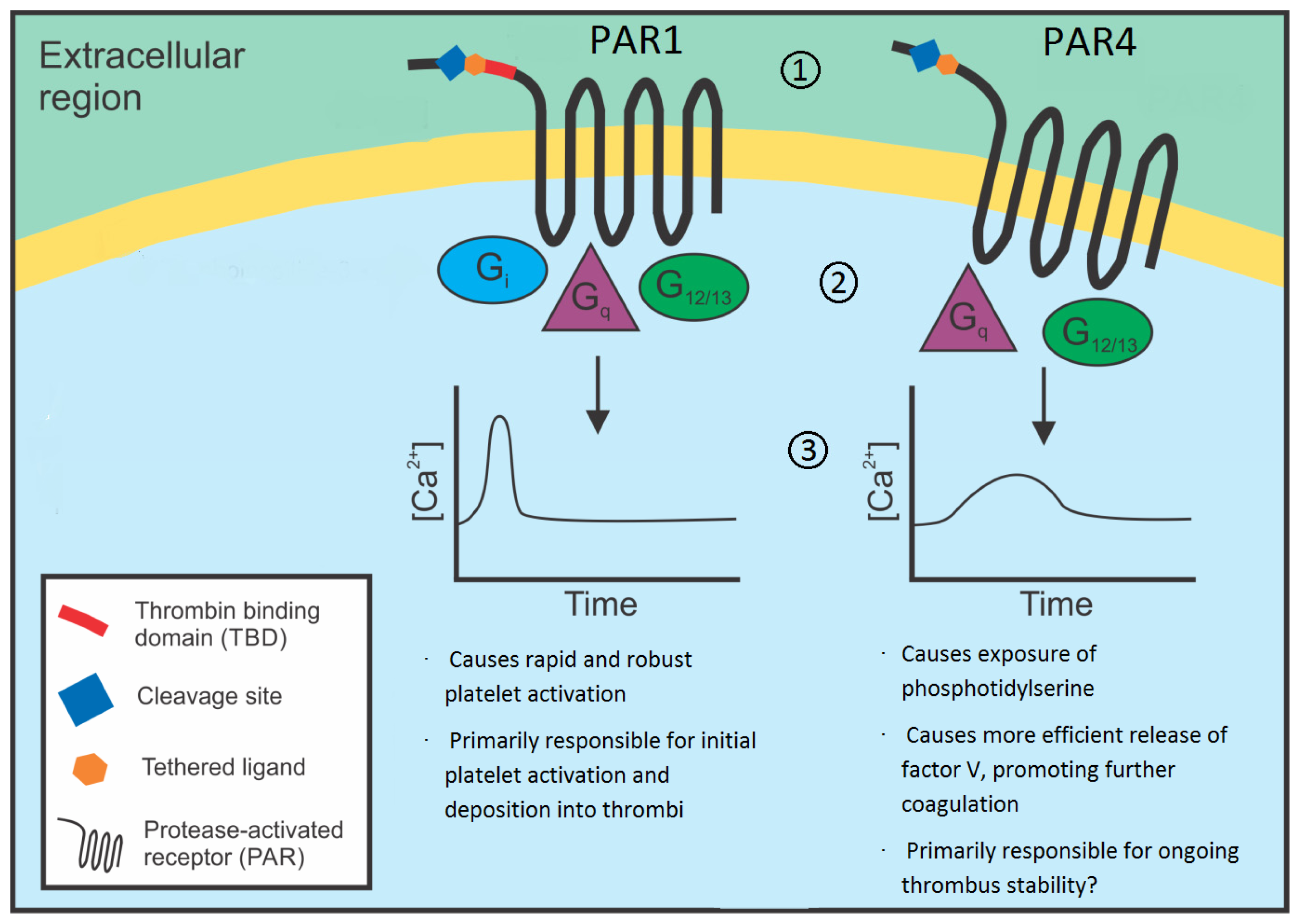
2.1. Distinct Mechanisms of Receptor Activation
2.2. Differences in Receptor Activation Mechanisms Promote the Targeting of PAR1 for the Prevention of Arterial Thrombosis
2.3. Differences in Receptor Activation Mechanisms Promote the Targeting of PAR2 for the Prevention of Histamine-Independent Itch
3. Exploiting Differences in PAR-Mediated Signaling for Therapeutic Benefit
3.1. Distinct Intracellular Signaling Events
3.2. Will Differences in Intracellular Signaling Events Promote the Targeting of PAR4 for the Prevention of Arterial Thrombosis?
3.3. Will Differences in Effector Mechanisms Promote the Selective Targeting of PAR1-Mediated Signaling for the Prevention of Vascular Inflammation?
4. Exploiting Differences in PAR-PAR Interactions for Therapeutic Benefit
4.1. Distinct Receptor Dimerization Events
4.2. Targeting PAR1-PAR4 Heterodimers for the Prevention of Arterial Thrombosis
4.3. Targeting PAR1-PAR2 Heterodimers for the Treatment of Sepsis
4.4. Targeting PAR3 Heterodimers for Therapeutic Benefit
5. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsT.S.S. and S.L.F researched and wrote the manuscript with equal contribution. J.R.H. supervised the work and wrote the manuscript.
References
- Kahn, M.L.; Zheng, Y.W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V., Jr.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar]
- Kahn, M.L.; Nakanishi-Matsui, M.; Shapiro, M.J.; Ishihara, H.; Coughlin, S.R. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Investig 1999, 103, 879–887. [Google Scholar]
- Vu, T.K.H.; Wheaton, V.I.; Hung, D.T.; Charo, I.; Coughlin, S.R. Domains specifying thrombin-receptor interaction. Nature 1991, 353, 674–677. [Google Scholar]
- Hung, D.T.; Wong, Y.H.; Vu, T.K.H.; Coughlin, S.R. The cloned platelet thrombin receptor couples to at least two distinct effectors to stimulate phosphoinositide hydrolysis and inhibit adenylyl cyclase. J. Biol. Chem 1992, 267, 20831–20834. [Google Scholar]
- Vu, T.K.H.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar]
- Scarborough, R.M.; Naughton, M.A.; Teng, W.; Hung, D.T.; Rose, J.; Vu, T.K.H.; Wheaton, V.I.; Turck, C.W.; Coughlin, S.R. Tethered ligand agonist peptides. Structural requirements for thrombin receptor activation reveal mechanism of proteolytic unmasking of agonist function. J. Biol. Chem 1992, 267, 13146–13149. [Google Scholar]
- Nystedt, S.; Emilsson, K.; Wahlestedt, C.; Sundelin, J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9208–9212. [Google Scholar]
- Camerer, E.; Huang, W.; Coughlin, S.R. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc. Natl. Acad. Sci. USA 2000, 97, 5255–5260. [Google Scholar]
- Kuliopulos, A.; Covic, L.; Seeley, S.K.; Sheridan, P.J.; Helin, J.; Costello, C.E. Plasmin desensitization of the PAR1 thrombin receptor: Kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry 1999, 38, 4572–4585. [Google Scholar]
- Riewald, M.; Petrovan, R.J.; Donner, A.; Mueller, B.M.; Ruf, W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science (N. Y.) 2002, 296, 1880–1882. [Google Scholar]
- Riewald, M.; Petrovan, R.J.; Donner, A.; Ruf, W. Activated protein C signals through the thrombin receptor PAR1 in endothelial cells. J. Endotoxin Res 2003, 9, 317–321. [Google Scholar]
- Molino, M.; Barnathan, E.S.; Numerof, R.; Clark, J.; Dreyer, M.; Cumashi, A.; Hoxie, J.A.; Schechter, N.; Woolkalis, M.; Brass, L.F. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J. Biol. Chem 1997, 272, 4043–4049. [Google Scholar]
- Wilson, S.; Greer, B.; Hooper, J.; Zijlstra, A.; Walker, B.; Quigley, J.; Hawthorne, S. The membrane-anchored serine protease, TMPRSS2, activates PAR-2 in prostate cancer cells. Biochem. J 2005, 388, 967–972. [Google Scholar]
- Takeuchi, T.; Harris, J.L.; Huang, W.; Yan, K.W.; Coughlin, S.R.; Craik, C.S. Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem 2000, 275, 26333–26342. [Google Scholar]
- Sambrano, G.R.; Huang, W.; Faruqi, T.; Mahrus, S.; Craik, C.; Coughlin, S.R. Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem 2000, 275, 6819–6823. [Google Scholar]
- Nakanishi-Matsui, M.; Zheng, Y.W.; Sulciner, D.J.; Weiss, E.J.; Ludeman, M.J.; Coughlin, S.R. PAR3 is a cofactor for PAR4 activation by thrombin. Nature 2000, 404, 609–613. [Google Scholar]
- Freynhofer, M.K.; Bruno, V.; Wojta, J.; Huber, K. The role of platelets in athero-thrombotic events. Curr. Pharm. Des 2012, 18, 5197–5214. [Google Scholar]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics-2013 update: A Report from the American Heart Association. Circulation 2013, 127, e6–e245. [Google Scholar]
- Hamilton, J.R. Protease-activated receptors as targets for antiplatelet therapy. Blood Rev 2009, 23, 61–65. [Google Scholar]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar]
- Morrow, D.A.; Braunwald, E.; Bonaca, M.P.; Ameriso, S.F.; Dalby, A.J.; Fish, M.P.; Fox, K.A.A.; Lipka, L.J.; Liu, X.; Nicolau, J.C.; et al. Vorapaxar in the secondary prevention of atherothrombotic events. N. Engl. J. Med 2012, 366, 1404–1413. [Google Scholar]
- Goto, S.; Ogawa, H.; Takeuchi, M.; Flather, M.D.; Bhatt, D.L. Double-blind, placebo-controlled Phase II studies of the protease-activated receptor 1 antagonist E5555 (atopaxar) in Japanese patients with acute coronary syndrome or high-risk coronary artery disease. Eur. Heart J 2010, 31, 2601–2613. [Google Scholar]
- Morrow, D.A.; Alberts, M.J.; Mohr, J.P.; Ameriso, S.F.; Bonaca, M.P.; Goto, S.; Hankey, G.J.; Murphy, S.A.; Scirica, B.M.; Braunwald, E. Efficacy and safety of vorapaxar in patients with prior ischemic stroke. Stroke 2013, 44, 691–698. [Google Scholar]
- Chatterjee, S.; Sharma, A.; Mukherjee, D. PAR-1 antagonists: Current state of evidence. J. Thromb. Thromb 2013, 35, 1–9. [Google Scholar]
- Tricoci, P.; Huang, Z.; Held, C.; Moliterno, D.J.; Armstrong, P.W.; van de Werf, F.; White, H.D.; Aylward, P.E.; Wallentin, L.; Chen, E.; et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N. Engl. J.Med 2012, 366, 20–33. [Google Scholar]
- Frungieri, M.B.; Weidinger, S.; Meineke, V.; Köhn, F.M.; Mayerhofer, A. Proliferative action of mast-cell tryptase is mediated by PAR2, COX2, prostaglandins, and PPARγ: Possible relevance to human fibrotic disorders. Proc. Natl. Acad. Sci. USA 2002, 99, 15072–15077. [Google Scholar]
- Akiyama, T.; Carstens, M.I.; Carstens, E. Enhanced scratching evoked by PAR-2 agonist and 5-HT but not histamine in a mouse model of chronic dry skin itch. Pain 2010, 151, 378–383. [Google Scholar]
- Akiyama, T.; Tominaga, M.; Davoodi, A.; Nagamine, M.; Blansit, K.; Horwitz, A.; Carstens, M.I.; Carstens, E. Cross-sensitization of histamine-independent itch in mouse primary sensory neurons. Neuroscience 2012, 226, 305–312. [Google Scholar]
- Steinhoff, M.; Neisius, U.; Ikoma, A.; Fartasch, M.; Heyer, G.; Skov, P.S.; Luger, T.A.; Schmelz, M. Proteinase-activated receptor-2 mediates itch: A novel pathway for pruritus in human skin. J. Neurosci 2003, 23, 6176–6180. [Google Scholar]
- Woulfe, D.S. Platelet G protein-coupled receptors in hemostasis and thrombosis. J. Thromb. Haemost 2005, 3, 2193–2200. [Google Scholar]
- Voss, B.; McLaughlin, J.N.; Holinstat, M.; Zent, R.; Hamm, H.E. PAR1, but not PAR4, activates human platelets through a G i/o/phosphoinositide-3 kinase signaling axis. Mol. Pharmacol 2007, 71, 1399–1406. [Google Scholar]
- Brass, L.F. Thrombin and platelet activation. Chest 2003, 124 Suppl 3, 18S–25S. [Google Scholar]
- Faruqi, T.R.; Weiss, E.J.; Shapiro, M.J.; Huang, W.; Coughlin, S.R. Structure-function analysis of protease-activated receptor 4 thetered Ligand peptides. Determinants of specificity and utility in assays of receptor function. J. Biol. Chem 2000, 275, 19728–19734. [Google Scholar]
- Kim, S.; Foster, C.; Lecchi, A.; Quinton, T.M.; Prosser, D.M.; Jin, J.; Cattaneo, M.; Kunapuli, S.P. Protease-activated receptors 1 and 4 do not stimulate Gi signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of Gi signaling. Blood 2002, 99, 3629–3636. [Google Scholar]
- Kanthou, C.; Kanse, S.M.; Kakkar, V.V.; Benzakour, O. Involvement of pertussis toxin-sensitive and -insensitive G proteins in α-thrombin signalling on cultured human vascular smooth muscle cells. Cell Signal 1996, 8, 59–66. [Google Scholar]
- Seminario-Vidal, L.; Kreda, S.; Jones, L.; O’Neal, W.; Trejo, J.; Boucher, R.C.; Lazarowski, E.R. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho- and Ca2+-dependent signaling pathways. J. Biol. Chem 2009, 284, 20638–20648. [Google Scholar]
- Ishihara, H.; Zeng, D.; Connolly, A.J.; Tam, C.; Coughlin, S.R. Antibodies to protease-activated receptor 3 inhibit activation of mouse platelets by thrombin. Blood 1998, 91, 4152–4157. [Google Scholar]
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386, 502–506. [Google Scholar]
- Babich, M.; King, K.L.; Nissenson, R.A. Thrombin stimulates inositol phosphate production and intracellular free calcium by a pertussus toxin-insensitive mechanism in osteosarcoma cells. Endocrinology 1990, 126, 948–954. [Google Scholar]
- Gao, L.; Smith, R.S.; Chen, L.M.; Chai, K.X.; Chao, L.; Chao, J. Tissue kallikrein promotes prostate cancer cell migration and invasion via a protease-activated receptor-1-dependent signaling pathway. Biol. Chem 2010, 391, 803–812. [Google Scholar]
- Nicole, O.; Goldshmidt, A.; Hamill, C.E.; Sorensen, S.D.; Sastre, A.; Lyuboslavsky, P.; Hepler, J.R.; McKeon, R.J.; Traynelis, S.F. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J. Neurosci 2005, 25, 4319–4329. [Google Scholar]
- Offermanns, S.; Laugwitz, K.L.; Spicher, K.; Schultz, G. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc. Natl. Acad. Sci. USA 1994, 91, 504–508. [Google Scholar]
- Brass, L.F.; Jiang, H.; Wu, J.; Stalker, T.J.; Zhu, L. Contact-dependent signaling events that promote thrombus formation. Blood Cells Mol. Dis 2006, 36, 157–161. [Google Scholar]
- McCoy, K.L.; Gyoneva, S.; Vellano, C.P.; Smrcka, A.V.; Traynelis, S.F.; Hepler, J.R. Protease-activated receptor 1 (PAR1) coupling to G q/11 but not to G i/o or G 12/13 is mediated by discrete amino acids within the receptor second intracellular loop. Cell Signal 2012, 24, 1351–1360. [Google Scholar]
- McCoy, K.L.; Traynelis, S.F.; Hepler, J.R. PAR1 and PAR2 couple to overlapping and distinct sets of G proteins and linked signaling pathways to differentially regulate cell physiology. Mol. Pharmacol 2010, 77, 1005–1015. [Google Scholar]
- Shapiro, M.J.; Weiss, E.J.; Faruqi, T.R.; Coughlin, S.R. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J. Biol. Chem 2000, 275, 25216–25221. [Google Scholar]
- Andersen, H.; Greenberg, D.L.; Fujikawa, K.; Xu, W.; Chung, D.W.; Davie, E.W. Protease-activated receptor 1 is the primary mediator of thrombin- stimulated platelet procoagulant activity. Proc. Natl. Acad. Sci. USA 1999, 96, 11189–11193. [Google Scholar]
- Weitz, J.I.; Crowther, M. Direct thrombin inhibitors. Thromb. Res 2002, 106, V275–V284. [Google Scholar]
- Duvernay, M.; Young, S.; Gailani, D.; Schoenecker, J.; Hamm, H.E. Protease-activated receptor (PAR) 1 and PAR4 differentially regulate factor V expression from human platelets. Mol. Pharmacol 2013, 83, 781–792. [Google Scholar]
- Vandendries, E.R.; Hamilton, J.R.; Coughlin, S.R.; Furie, B.; Furie, B.C. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc. Natl. Acad. Sci. USA 2007, 104, 288–292. [Google Scholar]
- Young, S.E.; Duvernay, M.T.; Schulte, M.L.; Lindsley, C.W.; Hamm, H.E. Synthesis of indole derived protease-activated receptor 4 antagonists and characterization in human platelets. PLoS One 2013, 8, e65528. [Google Scholar]
- Edelstein, L.C.; Simon, L.M.; Montoya, R.T.; Holinstat, M.; Chen, E.S.; Bergeron, A.; Kong, X.; Nagalla, S.; Mohandas, N.; Cohen, D.E.; et al. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat. Med 2013, 19, 1609–1616. [Google Scholar]
- Soh, U.J.K.; Trejo, J. Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. USA 2011, 108, E1372–E1380. [Google Scholar]
- Mosnier, L.O.; Sinha, R.K.; Burnier, L.; Bouwens, E.A.; Griffin, J.H. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 2012, 120, 5237–5246. [Google Scholar]
- Schulte, G.; Shenoy, S.K. β-Arrestin and dishevelled coordinate biased signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 19839–19840. [Google Scholar]
- Mihara, K.; Ramachandran, R.; Renaux, B.; Saifeddine, M.; Hollenberg, M.D. Neutrophil elastase and proteinase-3 trigger G proteinbiased signaling through proteinase-activated receptor-1 (PAR1). J. Biol. Chem 2013, 288, 32979–32990. [Google Scholar]
- Ramachandran, R.; Mihara, K.; Mathur, M.; Rochdi, M.D.; Bouvier, M.; DeFea, K.; Hollenberg, M.D. Agonist-biased signaling via proteinase activated receptor-2: Differential activation of calcium and mitogen-activated protein kinase pathways. Mol. Pharm 2009, 76, 791–801. [Google Scholar]
- Bouwens, E.A.M.; Stavenuiter, F.; Mosnier, L.O. Mechanisms of anticoagulant and cytoprotective actions of the protein C pathway. J. Thromb. Haemost 2013, 11 Suppl 1, 242–253. [Google Scholar]
- Feistritzer, C.; Schuepbach, R.A.; Mosnier, L.O.; Bush, L.A.; di Cera, E.; Griffin, J.H.; Riewald, M. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J. Biol. Chem 2006, 281, 20077–20084. [Google Scholar]
- Riewald, M.; Ruf, W. Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J. Biol. Chem 2005, 280, 19808–19814. [Google Scholar]
- Schuepbach, R.A.; Feistritzer, C.; Brass, L.F.; Riewald, M. Activated protein C cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood 2008, 111, 2667–2673. [Google Scholar]
- Zlokovic, B.V.; Griffin, J.H. Cytoprotective protein C pathways and implications for stroke and neurological disorders. Trends Neurosci 2011, 34, 198–209. [Google Scholar]
- Offermanns, S.; Toombs, C.F.; Hu, Y.H.; Simon, M.I. Defective platelet activation in Gα(q)-deficient mice. Nature 1997, 389, 183–186. [Google Scholar]
- Vouret-Craviari, V.; Boquet, P.; Pouysségur, J.; van Obberghen-Schilling, E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: Role of Rho proteins in endothelial barrier function. Mol. Biol. Cell 1998, 9, 2639–2653. [Google Scholar]
- Niessen, F.; Furlan-Freguia, C.; Fernandez, J.A.; Mosnier, L.O.; Castellino, F.J.; Weiler, H.; Rosen, H.; Griffin, J.H.; Ruf, W. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood 2009, 113, 2859–6286. [Google Scholar]
- Feistritzer, C.; Riewald, M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood 2005, 105, 3178–3184. [Google Scholar]
- Ludeman, M.J.; Kataoka, H.; Srinivasan, Y.; Esmon, N.L.; Esmon, C.T.; Coughlin, S.R. PAR1 cleavage and signaling in response to activated protein C and thrombin. J. Biol. Chem 2005, 280, 13122–13128. [Google Scholar]
- Camerer, E.; Cornelissen, I.; Kataoka, H.; Duong, D.N.; Zheng, Y.W.; Coughlin, S.R. Roles of protease-activated receptors in a mouse model of endotoxemia. Blood 2006, 107, 3912–3921. [Google Scholar]
- Kaneider, N.C.; Leger, A.J.; Agarwal, A.; Nguyen, N.; Perides, G.; Derian, C.; Covic, L.; Kuliopulos, A. “Role reversal” for the receptor PAR1 in sepsis-induced vascular damage. Nat. Immunol 2007, 8, 1303–1312. [Google Scholar]
- Pawlinski, R.; Pedersen, B.; Schabbauer, G.; Tencati, M.; Holscher, T.; Boisvert, W.; Andrada-Gordon, P.; Frank, R.D.; Mackman, N. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood 2004, 103, 1342–1347. [Google Scholar]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal 2013, 11. [Google Scholar] [CrossRef]
- Lin, H.; Liu, A.P.; Smith, T.H.; Trejo, J. Cofactoring and dimerization of proteinase-activated receptors. Pharmacol. Rev 2013, 65, 1198–1213. [Google Scholar]
- Blackhart, B.D.; Emilsson, K.; Nguyen, D.; Teng, W.; Martelli, A.J.; Nystedt, S.; Sundelin, J.; Scarborough, R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem 1996, 271, 16466–16471. [Google Scholar]
- O’Brien, P.J.; Prevost, N.; Molino, M.; Hollinger, M.K.; Woolkalis, M.J.; Woulfe, D.S.; Brass, L.F. Thrombin responses in human endothelial cells: Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J. Biol. Chem 2000, 275, 13502–13509. [Google Scholar]
- Leger, A.J.; Jacques, S.L.; Badar, J.; Kaneider, N.C.; Derian, C.K.; Andrade-Gordon, P.; Covic, L.; Kuliopulos, A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation 2006, 113, 1244–1254. [Google Scholar]
- Covic, L.; Misra, M.; Badar, J.; Singh, C.; Kuliopulos, A. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat. Med 2002, 8, 1161–1165. [Google Scholar]
- De la Fuente, M.; Noble, D.N.; Verma, S.; Nieman, M.T. Mapping human protease-activated receptor 4 (PAR4) homodimer interface to transmembrane helix 4. J. Biol. Chem 2012, 287, 10414–10423. [Google Scholar]
- McLaughlin, J.N.; Patterson, M.M.; Malik, A.B. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc. Natl. Acad. Sci. USA 2007, 104, 5662–5667. [Google Scholar]
- Madhusudhan, T.; Wang, H.; Straub, B.K.; Grone, E.; Zhou, Q.; Shahzad, K.; Muller-Krebs, S.; Schwenger, V.; Gerlitz, B.; Grinnell, B.W.; et al. Cytoprotective signaling by activated protein C requires protease-activated receptor-3 in podocytes. Blood 2012, 119, 874–883. [Google Scholar]

© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sidhu, T.S.; French, S.L.; Hamilton, J.R. Differential Signaling by Protease-Activated Receptors: Implications for Therapeutic Targeting. Int. J. Mol. Sci. 2014, 15, 6169-6183. https://doi.org/10.3390/ijms15046169
Sidhu TS, French SL, Hamilton JR. Differential Signaling by Protease-Activated Receptors: Implications for Therapeutic Targeting. International Journal of Molecular Sciences. 2014; 15(4):6169-6183. https://doi.org/10.3390/ijms15046169
Chicago/Turabian StyleSidhu, Tejminder S., Shauna L. French, and Justin R. Hamilton. 2014. "Differential Signaling by Protease-Activated Receptors: Implications for Therapeutic Targeting" International Journal of Molecular Sciences 15, no. 4: 6169-6183. https://doi.org/10.3390/ijms15046169