2.1. Solubility and Intrinsic Dissolution Ratio of NCE
The equilibrated solubility of NCE in phosphate buffers was shown in
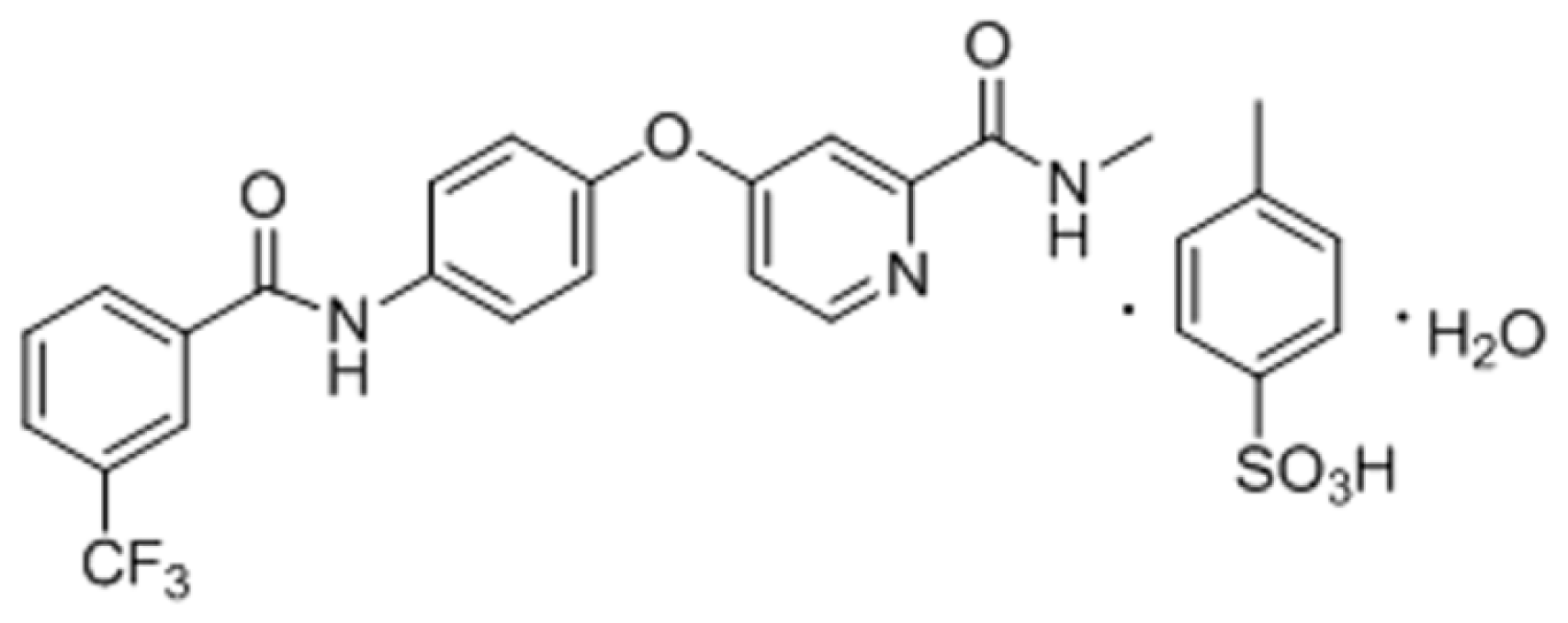
Table 1. The aqueous solubility of NCE over the pH range of 1.0–7.4 was quite low with a maximum solubility of 81.73 μg/mL at pH 1.0. From its chemical structure (
Figure 1), the parent structure of NCE is apparently a weak base and formulated as a methylbenzene sulfonate salt. The p
Ka value of its conjugate acid has been determined as 2.86, which is attributed to the pyridine group (p
Ka = 5.25 [
13]). Thus, NCE would be ionized at low pH (pH = 1.0) with a high solubility and a mainly molecular state at high pH (such as 5.0, 6.8 and 7.4) with a low solubility. According to the Henderson-Hasselbach equation [
14], NCE would maintain unionized within the pH range of GI tract. The partition coefficient (log
P value) was calculated with 3.1 ± 0.04 in 0.1 M HCl solution (pH = 1.0) and 4.20 ± 0.07 in phosphate buffer solution (PBS) 6.8 solution, indicating that NCE is highly lipid soluble and could be well absorbed
in vivo.
Intrinsic dissolution ratio is defined as the dissolution rate of a pure drug substance under the condition of constant surface area, agitation or stirring speed, pH and ionic strength of the dissolution medium. As a rate characterization, IDR data has a good correlation with
in vivo drug dissolution rate [
15]. Because of the low solubility of NCE in the tested phosphate buffers, sodium dodecyl sulfate (SDS) was added to increase the solubility to meet the sink condition which refers that the concentration of drug in release medium is far smaller than its saturation concentration. Three different SDS concentrations were tested. The solubilities of NCE were 54.46 (0.4% SDS), 131.83 (1% SDS) and 255.91 (2% SDS) μg/mL. The volume of the release medium in this experiment was 900 mL, thus the introduction of 1% SDS into the medium was enough to achieve sink condition.
The IDR was estimated by the dissolution data from the initial 4 h because it got close to the saturate concentration after 4 h with a decreased dissolution rate (
Figure 2). The IDR of NCE was calculated to be 1 × 10
−4 mg·min
−1·cm
−2, which was below the borderline value of 1 mg·min
−1·cm
−2 [
15,
16]. Therefore, NCE was classified as a compound with low solubility.
2.3. The Effect of Intestinal Site and NCE Concentration on Intestinal Permeability
In vitro and
in vivo methods are frequently used to predict the drug absorption in small intestine [
17]. Furthermore,
in vivo method was significantly superior to
in vitro method because there was no damange to the lymphatic and circulatory systems of the studied tissues. Among the
in vivo methods, there are
in vivo circulation methods and single pass intestine perfusion (SPIP) methods. As for
in vivo circulation method, the long-term experiment process and high perfusion speed can cause damage to the intestinal mucosa, leading to an increase in drug absorption. Thus, the measured value and the true value have a large deviation. SPIP approach is the most frequently used method that provides
in situ experimental conditions simulating oral administration [
16,
18–
20]. By possessing a preserved microenvironment around the intestinal membrane, SPIP method is able to accurately characterize the drug absorption [
21] in different small intestine regions. In addition, the data have a high correlation with the absorption characteristics in human beings [
11]. The effective permeability (
Peff) values (effective permeability) of human and rats had been proved to be highly relevant [
16,
22]. Therefore, the permeability investigation in rat intestine was necessary, which could be used to predict the oral absorption of NCE in human.
Several methods can be applied to calculate effective permeability of NCE in rat intestine. Such as the gravimetric method, the phenol red or
14C-PEG marked ways to indicate the change of perfusion volume [
23]. However, the phenol red itself can be absorbed partly by the intestine. Also, it can affect the intestinal transport and analysis of certain compounds. The
14C-PEG marker exits radioactivity and security issues. Thus, gravimetric method was chosen to be applied in this study.
As previously described, due to the poor solubility of NCE in the perfusion buffer, different concentrations of SDS (0.4%, 1% and 2%,
w/
v) were added into the perfusion buffer to improve the solubility of NCE. The concentration of NCE in perfusion buffer containing 0.4% SDS was high enough to conduct the following intestinal permeability study. Additionally, according to preliminary experiments, SDS with concentrations higher than 1% would be toxic to rats [
24]. Thus, 0.4% SDS was finally chosen to be added into the final perfusion buffer solution to improve the solubility of NCE.
In order to investigate whether 0.4% SDS has any effect on intestinal absorption of NCE, 2 μg/mL of NCE with and without 0.4% SDS was applied to three intestinal segments using the rat perfusion model in this study. The effective permeability of NCE in duodenum, jejunum and ileum was shown in
Table 3. The
Peff values of NCE with and without SDS in any intestinal segment were similar (
p > 0.05). No significant enhancement or reduction absorption was found in any intestinal segments, indicating that 0.4% SDS had no effects on intestinal absorption of NCE.
To achieve a more comprehensive evaluation of drug permeability, a high permeability compound (theophylline) and a low permeability compound (ranitidine) were applied in this study [
8,
25]. The absorption mechanism of these two compounds was passive diffusion [
16,
26,
27], suggesting that the drug concentration had no effects on its absorption. Thus, 40 μg/mL of theophylline, ranitidine and NCE perfusion solution were prepared for the SPIP study.
The effective permeability (
Peff) of NCE, theophylline and ranitidine in duodenum, jejunum and ileum was shown in
Tables 3 and
4. As shown in
Table 4, significant difference was found among these three drugs in the same intestinal segment (
p < 0.05). The
Peff value of NCE was higher than ranitidine and lower than theophylline in any intestinal segment. As described previously, theophylline was a high permeability drug and ranitidine was a low permeability drug [
25]. The
Peff values of NCE were more proximate to that of theophylline with high permeability, suggesting that NCE has a high permeability. The permeability coefficients of NCE obtained from SPIP study in three intestinal segments at three different concentrations, with the minimum of
Peff (0.20 × 10
−4 cm/s) found in jejunum and the maximal
Peff (0.56 × 10
−4 cm/s) found in duodenum, were comparable to propranolol (
Peff of propranolol: 0.30–0.75 × 10
−4 cm/s) which was a widely used reference compound for high permeability evaluation [
4,
28]. Hence, NCE was considered to be well absorbed in the whole intestine and categorized as a high permeability drug. In conclusion, NCE was categorized as BCS class II drug due to its low solubility and high permeability. All the data would provide a reference for biopharmaceutics classification research of other novel drug candidates.
The effective permeability (
Peff)of different concentrations of NCE in duodenum, jejunum and ileum was shown in
Table 5. The
Peff values of NCE in different intestinal segment at the same concentration were similar (
p > 0.05). No significant intestinal site dependent absorption was found at all concentrations, indicating that NCE was absorbed in the whole intestine without any site specific absorption sites. In addition, when the concentration of NCE increased from 2–40 μg/mL, the
Peff values in the same intestinal segment had no significant difference (
p > 0.05). Generally, when the drug’s permeability does not change with the change of its concentrations, its transportation mechanism was passive diffusion according to Thomas J Cook [
29]. Hence, the transportation mechanism of NCE in intestine was passive diffusion.





{kind=link}
{kind=link}
{kind=link}