Cancer Stem Cells: Biological Functions and Therapeutically Targeting

{kind=link}
{kind=link}
Abstract
:1. Introduction
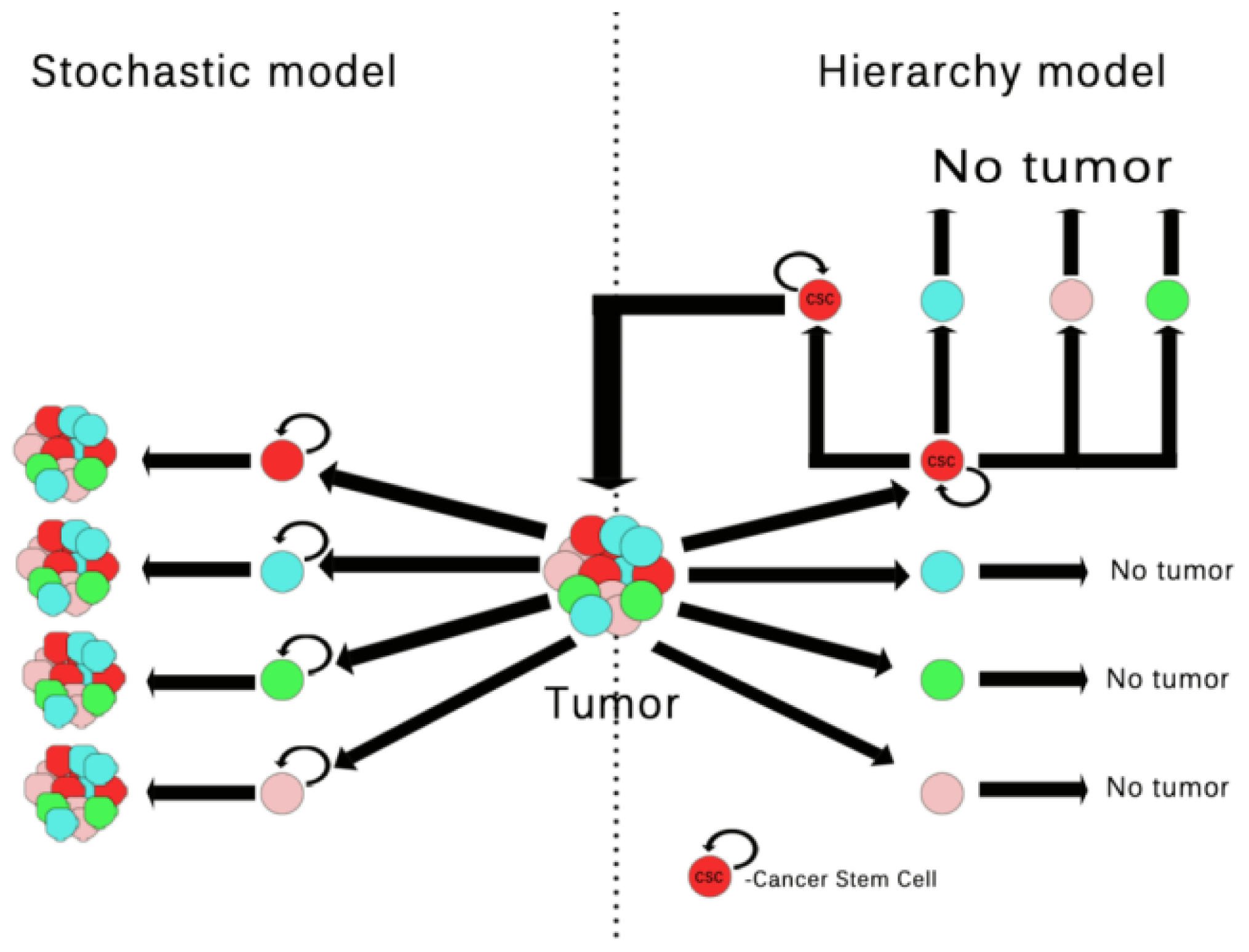
2. Tumor Cells vs. Tumor Stem Cells
3. Molecular Signalling Pathways in Cancer Stem Cells
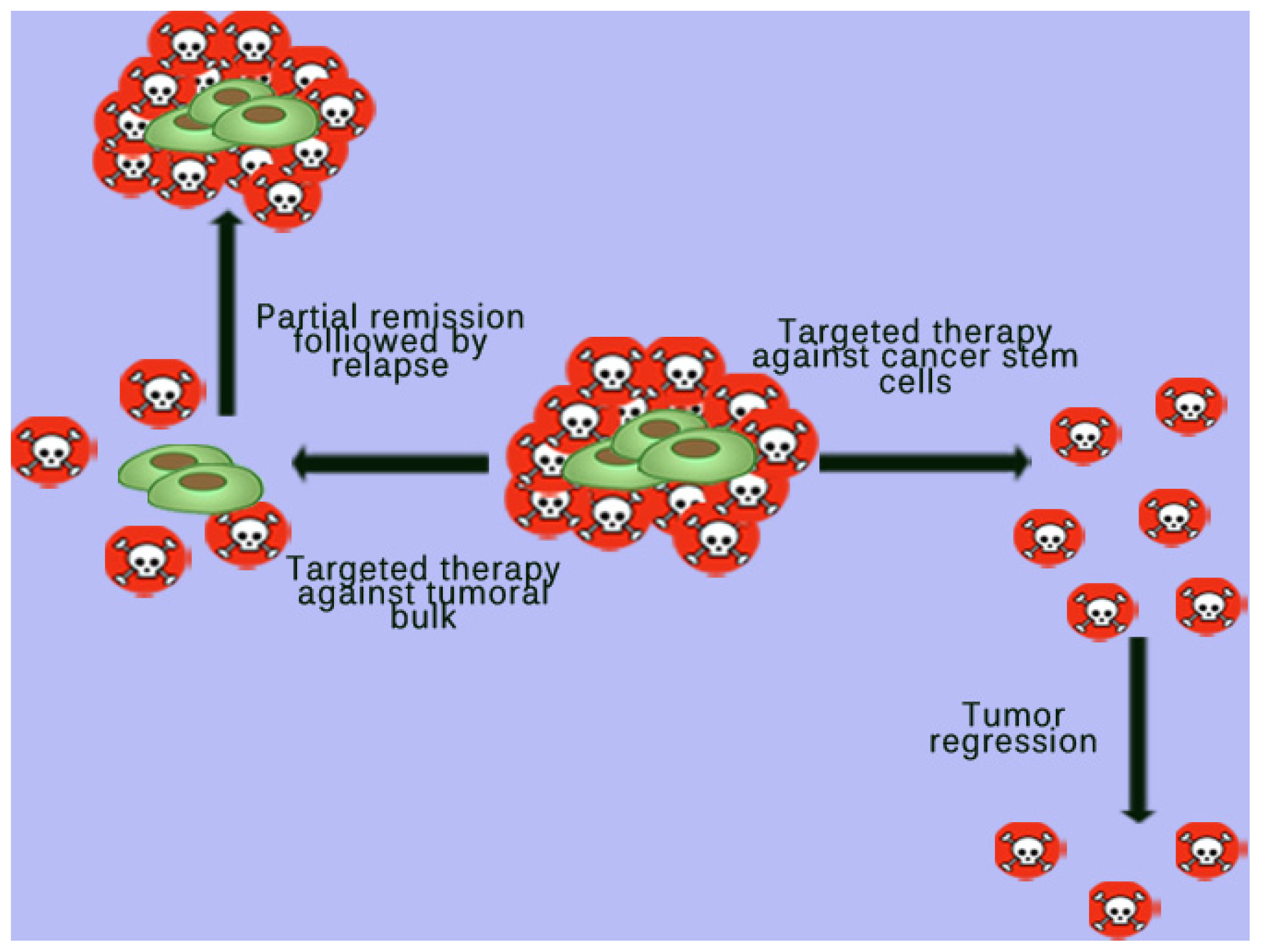
4. Targeted Therapy against Cancer Stem Cells (CSCs)
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793. [Google Scholar]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med 1997, 3, 730–737. [Google Scholar]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol 2004, 5, 738–743. [Google Scholar]
- Passegué, E.; Jamieson, C.H.M.; Ailles, L.E.; Weissman, I.L. Normal and leukemic hematopoiesis: Are leukemias a stem cell disorder or a reacquisition of stem cell characteristics? Proc. Natl. Acad. Sci. USA 2003, 100, 11842–11849. [Google Scholar]
- Castelo-Branco, P.; Zhang, C.; Lipman, T.; Fujitani, M.; Hansford, L.; Clarke, I.; Harley, C.B.; Tressler, R.; Malkin, D.; Walker, E.; et al. Neural tumor-initiating cells have distinct telomere maintenance and can be safely targeted for telomerase inhibition. Clin. Cancer Res 2011, 17, 111–121. [Google Scholar]
- Li, C.; Hynes, M.J.; Jing, J. Pancreatic cancer stem cells: New direction for pancreatic cancer treatment. Trends Bio/Pharm. Ind 2010, 6, 34–40. [Google Scholar]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68, 4311–4320. [Google Scholar]
- Miki, J.; Furusato, B.; Li, H.; Gu, Y.; Takahashi, H.; Egawa, S.; Sesterhenn, I.A.; McLeod, D.G.; Srivastava, S.; Rhim, J.S. Identification of putative stem cell markers, CD133 and CXCR4, in hTERT-immortalized primary nonmalignant and malignant tumor-derived human prostate epithelial cell lines and in prostate cancer specimens. Cancer Res 2007, 67, 3153–3161. [Google Scholar]
- Beier, D.; Hau, P.; Proescholdt, M.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 2007, 67, 4010–4015. [Google Scholar]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar]
- Tirino, V.; Desiderio, V.; Paino, F.; de Rosa, A.; Papaccio, F.; Fazioli, F.; Pirozzi, G.; Papaccio, G. Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. FASEB J. 2011, 6, 2022–2030. [Google Scholar]
- Suvà, M.L.; Riggi, N.; Stehle, J.C.; Baumer, K.; Tercier, S.; Joseph, J.M.; Suva, D.; Clement, V.; Provero, P.; Cironi, L.; et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res 2009, 69, 1776–1781. [Google Scholar]
- Boveri, T. The Origin of Malignant Tumors; Williams and Wilkins: Baltimore, MD, USA, 1914. [Google Scholar]
- Tyzzer, E.E. Tumor immunity. J. Cancer Res 1916, 1, 125–156. [Google Scholar]
- Muller, H.J. Radiation damage to the genetic material. Sci. Prog 1951, 7, 33–59. [Google Scholar]
- Berenblum, I.; Shubik, P. A new, quantitative, approach to the study of the stages of chemical carcinogenesis in the mouse’s skin. Br. J. Cancer 1947, 1, 383–391. [Google Scholar]
- Moolgavkar, S.H.; Knudson, A.G. Mutation and cancer: A model for human carcinogenesis. J. Natl. Cancer Inst 1981, 66, 1037–1052. [Google Scholar]
- Knudson, A.G., Jr. Hereditary cancers of man. Cancer Investig 1983, 1, 187–193. [Google Scholar]
- Knudson, A.G., Jr. Hereditary cancer, oncogene, and antioncogenes. Cancer Res 1985, 45, 1437–1443. [Google Scholar]
- Harbour, J.W.; Lai, S.L.; Whang-Peng, J.; Gazdar, A.F.; Minna, J.D.; Kaye, F.J. Abnormalities in structure and expression of the human retinoblastoma gene in SCLC. Science 1988, 24, 353–357. [Google Scholar]
- Lee, W.H.; Shew, J.Y.; Hong, F.D.; Sery, T.W.; Donoso, L.A.; Young, L.J.; Bookstein, R.; Lee, E.Y. The retinoblastoma susceptibility gene encodes a nuclear phosphoprotein associated with DNA binding activity. Nature 1987, 329, 642–645. [Google Scholar]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar]
- Hanahan, D.; Weinberg, R. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Weinberg, R.A. The Biology of Cancer; Taylor & Francis: New York, NY, USA, 2006. [Google Scholar]
- Triolo, V.A. Nineteenth century foundations of cancer research advances in tumor pathology, nomenclature, and theories of oncogenesis. Cancer Res 1965, 25, 76–98. [Google Scholar]
- Soto, A.M.; Sonnenschein, C. Emergentism as a default: Cancer as a problem of tissue organization. J. Biosci 2005, 30, 103–118. [Google Scholar]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet 2006, 7, 21–33. [Google Scholar]
- Rock, J.R.; Randell, S.H.; Hogan, B.L. Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech 2010, 3, 545–556. [Google Scholar]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar]
- Van Keymeulen, A.; Blanpain, C. Tracing epithelial stem cells during development, homeostasis, and repair. J. Cell Biol 2012, 197, 575–584. [Google Scholar]
- Moore, M.A.; Williams, N.; Metcalf, D. In vitro colony formation by normal and leukemic human hematopoietic cells: Characterization of the colony forming cells. J. Nat. Cancer. Inst 1973, 50, 603–623. [Google Scholar]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Ann. Rev. Med 2007, 58, 267–284. [Google Scholar]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells vs. clonal evolution. Cell 2009, 138, 822–829. [Google Scholar]
- Dick, J.E. Breast cancer stem cells revealed. Proc. Natl. Acad. Sci. USA 2003, 100, 3547–3549. [Google Scholar]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res 66.
- Sell, S. On the stem cell origin of cancer. Am. J. Pathol 2010, 176, 2584–2594. [Google Scholar]
- Pierce, G.B.; Dixon, F.J. The demonstration of teratogenesis by metamorphosis of multipotential cells. Cancer 1959, 12, 573–583. [Google Scholar]
- Ushijima, T. Epigenetic field for cancerization. J. Biochem. Mol. Biol 2007, 40, 142–150. [Google Scholar]
- Ushijima, T. Detection and interpretation of altered methylation patterns in cancer cells. Nat. Rev. Cancer 2005, 5, 223–231. [Google Scholar]
- Sell, S. Stem cells in hepato carcinogenesis. Cell Sci. Rev 2006, 3, 1742–8130. [Google Scholar]
- Rous, P. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J. Exp. Med 1911, 13, 397–411. [Google Scholar]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar]
- Odoux, C.; Fohrer, H.; Hoppo, T.; Guzik, L.; Stolz, D.B.; Lewis, D.W.; Gollin, S.M.; Gamblin, T.C.; Geller, D.A.; Lagasse, E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res 2008, 68, 6932–6941. [Google Scholar]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar]
- Bapat, S.A. Evolution of cancer stem cells. Semin. Cancer Biol 2007, 17, 204–213. [Google Scholar]
- Chaffer, C.L.; Brueckmann, I.; Scheela, C. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar]
- Dick, J.E. Looking ahead in cancer stem cell research. Nat. Biotechnol 2009, 27, 44–46. [Google Scholar]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumor formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar]
- Dingli, D.; Traulsen, A.; Pacheco, J.M. Stochastic dynamics of hematopoietic tumor stem cells. Cell Cycle 2007, 6, 461–466. [Google Scholar]
- Vermeulen, L.; de Sousa, E.; Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol 2010, 12, 468–476. [Google Scholar]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Galorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res 2010, 70, 6945–6956. [Google Scholar]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; de Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 2012, 72, 5130–5140. [Google Scholar]
- Fiaschi, T.; Giannoni, E.; Taddei, M.L.; Cirri, P.; Marini, A.; Pintus, G.; Nativi, C.; Richichi, B.; Scozzafava, A.; Carta, F.; et al. Carbonic anhydrase IX from cancer associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle 2013, 12, 1791–1801. [Google Scholar]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat 1995, 154, 8–20. [Google Scholar]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol 2006, 7, 131–142. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar]
- Giannoni, E.; Bianchini, F.; Calorini, L.; Chiarugi, P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid. Redox. Signal 2011, 14, 2361–2371. [Google Scholar]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumor cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumor-initiating cells. Nature 2006, 444, 761–765. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D. Identification of human brain tumor initiating cells. Nature 2004, 432, 396–401. [Google Scholar]
- Cho, R.W.; Clarke, M.F. Recent advances in cancer stem cells. Curr. Opin. Genet. Dev 2008, 18, 48–53. [Google Scholar]
- Bloushtain-Qimron, N.; Yao, J.; Snyder, E.L.; Shipitsin, M.; Campbell, L.L.; Mani, S.A. Cell type-specific DNA methylation patterns in the human breast. Proc. Natl. Acad. Sci. USA 2008, 105, 14076–14081. [Google Scholar]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar]
- Kondo, T.; Setoguchi, T.; Taga, T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc. Natl. Acad. Sci. USA 2004, 101, 781–786. [Google Scholar]
- Zheng, X.; Shen, G.; Yang, X.; Liu, W. Most C6 cells are cancer stem cells: Evidence from clonal and population analyses. Cancer Res 2007, 67, 3691–3697. [Google Scholar]
- Fukaya, R.; Ohta, S.; Yamaguchi, M.; Fujii, H.; Kawakami, Y. Isolation of cancer stem-like cells from a side population of a human glioblastoma cell line, SK-MG-1. Cancer Lett 2010, 291, 150–157. [Google Scholar]
- Qiang, L.; Yang, Y.; Ma, Y.J.; Chen, F.H.; Zhang, L.B. Isolation and characterization of cancer stem like cells in human glioblasoma cell lines. Cancer Lett. 2009, 279, 13–21. [Google Scholar]
- Bale, A.E.; Yu, K.P. The hedgehog pathway and basal cell carcinomas. Hum. Mol. Genet 2001, 10, 757–762. [Google Scholar]
- Sheng, T.; Li, C.; Zhang, X.; Chi, S.; He, N.; Chen, K.; McCormick, F.; Gatalica, Z.; Xie, J. Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer 2004, 3, 29. [Google Scholar]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol 2011, 8, 97–106. [Google Scholar]
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signalling in cancer. Oncogene 2008, 27, 5124–5131. [Google Scholar]
- Ma, X.; Chen, K.; Huang, S.; Zhang, X.; Adegboyega, P.A.; Evers, B.M. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis 2005, 26, 1698–1705. [Google Scholar]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X. Mutations in SUFU predispose to medulloblastoma. Nat. Genet 2002, 31, 306–310. [Google Scholar]
- Polakis, P. Wnt signaling and cancer. Genes Dev 2000, 14, 1837–1851. [Google Scholar]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar]
- Rheinbay, E.; Suvà, M.L. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep 2013, 3, 1567–1579. [Google Scholar]
- Zhang, N.; Wei, P.; Gong, A. FoxM1 Promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res 2008, 68, 9125–9130. [Google Scholar]
- Beà, S.; Tort, F.; Pinyol, M.; Puig, X.; Hernández, L.; Hernández, S. BMI-1 gene amplification and over-expression in hematological malignancies occur mainly in mantle cell lymphomas. Cancer Res 2001, 61, 2409–2412. [Google Scholar]
- Song, L.B.; Zeng, M.S.; Liao, W.T.; Zhang, L.; Mo, H.Y.; Liu, W.L. Bmi-1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res 2006, 66, 6225–6232. [Google Scholar]
- Bhattacharya, R.; Nicoloso, M.; Arvizo, R.; Wang, E.; Cortez, A.; Rossi, S. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res 2009, 69, 9090–9095. [Google Scholar]
- Lowell, E.M.; Westerman, B.A.; Ermilov, A.N. BMI1 is required for Hedgehog pathway—Driven medulloblastoma expansion. Neoplasia 2008, 10, 1343–1349. [Google Scholar]
- Fujiwara, Y.; Yoshikawa, R.; Tao, L.; Tsujimura, T.; Sasako, M. Stemness signature of BMI1 and clinical outcome in esophageal cancer patients undergoing neoadjuvant chemoradiotherapy. J. Clin. Oncol 2009, 27, 4572. [Google Scholar]
- Jiang, L.; Li, J.; Song, L. Bmi-1, stem cells and cancer. Acta Biochim. Biophys. Sin 2009, 41, 527–534. [Google Scholar]
- Cleton-Jansen, A.M.; Anninga, J.K.; Briaire-de Bruijn, I.H.; Romeo, S. Profiling of high-grade central osteosarcoma and its putative progenitor cells identifies tumorigenic pathways. Br. J. Cancer 2009, 101, 1909–1918. [Google Scholar]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X. Let-7 regulates self-renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar]
- Carapancea, M.; Alexandru, O.; Fetea, A.S.; Dragutescu, L.; Castro, J.; Georgescu, A.; Popa-Wagner, A. Growth factor receptors signalling in glioblastoma cells: Therapeutic implications. J. Neurooncol 2009, 92, 137–147. [Google Scholar]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: Insights into multidrug resistance and therapeutic development. Clin. Pharmacol. Ther 2011, 89, 491–502. [Google Scholar]
- Vermeulen, L.; de Sousa, E.; Melo, F.; Richel, D.J.; Medema, J.P. The developing cancer stem-cell model: Clinical challenges and opportunities. Lancet Oncol 2012, 13, e83–e89. [Google Scholar]
- Zhou, B.B.; Zhang, H.; Damelin, M.; Geles, K.G.; Grindley, J.C.; Dirks, P.B. Tumor-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov 2009, 8, 806–823. [Google Scholar]
- Naujokat, C.; Steinhart, R. Salinomycin as a drug for targeting human cancer stem cells. J. Biomed. Biotechnol 2012, 2012, 950658. [Google Scholar]
- Cordon-Cardo, C. Suppression of acquired docetaxel resistance in prostate cancer through depletion of Notch- and Hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar]
- Rosen, J.M.; Jordan, C.T. The increasing complexity of the cancer stem cell paradigm. Science 2009, 324, 1670. [Google Scholar]
- Katoh, M. Network of Wnt and other regulatory signalling cascades in pluripotent stem cells and cancer stem cells. Curr. Pharm. Biotechnol 2011, 12, 160–170. [Google Scholar]
- Zeng, G.; Apte, U.; Cieply, B. siRNA-mediated β-catenin knockdown in human hepatoma cells results in decreased growth and survival. Neoplasia 2007, 9, 951–959. [Google Scholar]
- Adler, E.M. Inhibiting Wnt signalling. Sci. Signal 2009, 2, ec326. [Google Scholar]
- Stanton, B.Z.; Peng, L.F. Small-molecule modulators of the Sonic Hedgehog signalling pathway. Mol. Biosyst 2010, 6, 44–54. [Google Scholar]
- Wang, Q.; Huang, S.; Yang, L.; Zhao, L.; Yin, Y.; Liu, Z. Down-regulation of sonic hedgehog signaling pathway activity is involved in 5-fluorouracil-induced apoptosis and motility inhibition in Hep3B cells. Acta Biochim. Biophys. Sin 2008, 40, 819–829. [Google Scholar]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W. EpCAM-positive hepatocellular carcinoma cells are tumor - initiating cells with stem/progenitor cell features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar]
- Chen, Y.; Yu, D.; Zhang, H.; He, H.; Zhang, C. CD133(+)EpCAM(+) phenotype possesses more characteristics of tumor initiating cells in hepatocellular carcinoma Huh7 cells. Int. J. Biol. Sci 2012, 8, 992–1004. [Google Scholar]
- Bin, B.; Asfar, A.; Amro, A.; Aamir, A. Pancreatic cancer stem-like cells display aggressive behavior mediated via activation of FoxQ1. J. Biol. Chem 2014. [Google Scholar] [CrossRef]
- Zhu, Z.; Hao, X.; Yan, M.; Yao, M.; Ge, C.; Gu, J.; Li, J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2067–2078. [Google Scholar]
- Kurtz, J.E.; Dufour, P. Adecatumumab: An anti-EpCAM monoclonal antibody, from the bench to the bedside. Expert Opin. Biol. Ther 2010, 10, 951–958. [Google Scholar]
- Motohashi, T.; Aoki, H.; Chiba, K. Multipotent cell fate of neural crest-like cells derived from embryonic stem cells. Stem Cells 2007, 25, 402–410. [Google Scholar]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res 2010, 16, 3113–3120. [Google Scholar]
- Alison, M.R.; Lim, S.M.; Nicholson, L.J. Cancer stem cells: Problems for therapy? J. Pathol 2010, 223, 148–162. [Google Scholar]
- Hermann, P.C.; Trabulo, S.M.; Sainz, B., Jr.; Balic, A.; Garcia, E.; Hahn, S.A. Multimodal treatment eliminates cancer stem cells and leads to long-term survival in primary human pancreatic cancer tissue xenografts. PLoS One 2013, 8, e66371. [Google Scholar]
- Chen, G.; Xu, S.; Renko, K.; Derwahl, M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. J. Clin. Endocrin. Metab 2012, 97, 1–11. [Google Scholar]
- Chan, C.H.; Morrow, J.K.; Li, C.F.; Gao, Y.; Jin, G.; Moten, A. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013, 154, 556–568. [Google Scholar]
- Alisi, A.; Cho, W.C.; Locatelli, F.; Fruci, D. Multidrug resistance and cancer stem cells in neuroblastoma and hepatoblastoma. Int. J. Mol. Sci 2013, 14, 24706–24725. [Google Scholar]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Ann. Rev. Med 2002, 53, 615–627. [Google Scholar]
- Ullah, M.F. Cancer multidrug resistance (MDR): A major impediment to effective chemotherapy. Asian Pac. J. Cancer Prev 2008, 9, 1–6. [Google Scholar]
- Science Daily. Available online: http://www.sciencedaily.com/releases/2012/09/120910122114.htm accessed on 10 October 2013.
- Ruck, P.; Xiao, J.C.; Pietsch, T.; von Schweinitz, D.; Kaiserling, E. Hepatic stem-like cells in hepatoblastoma: Expression of cytokeratin 7, albumin and oval cell associated antigens detected by OV-1 and OV-6. Histopathology 1997, 31, 324–329. [Google Scholar]


© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ciurea, M.E.; Georgescu, A.M.; Purcaru, S.O.; Artene, S.-A.; Emami, G.H.; Boldeanu, M.V.; Tache, D.E.; Dricu, A. Cancer Stem Cells: Biological Functions and Therapeutically Targeting. Int. J. Mol. Sci. 2014, 15, 8169-8185. https://doi.org/10.3390/ijms15058169
Ciurea ME, Georgescu AM, Purcaru SO, Artene S-A, Emami GH, Boldeanu MV, Tache DE, Dricu A. Cancer Stem Cells: Biological Functions and Therapeutically Targeting. International Journal of Molecular Sciences. 2014; 15(5):8169-8185. https://doi.org/10.3390/ijms15058169
Chicago/Turabian StyleCiurea, Marius Eugen, Ada Maria Georgescu, Stefana Oana Purcaru, Stefan-Alexandru Artene, Ghazaleh Hooshyar Emami, Mihai Virgil Boldeanu, Daniela Elise Tache, and Anica Dricu. 2014. "Cancer Stem Cells: Biological Functions and Therapeutically Targeting" International Journal of Molecular Sciences 15, no. 5: 8169-8185. https://doi.org/10.3390/ijms15058169