The Discovery of Potentially Selective Human Neuronal Nitric Oxide Synthase (nNOS) Inhibitors: A Combination of Pharmacophore Modelling, CoMFA, Virtual Screening and Molecular Docking Studies

Abstract
:
1. Introduction
2. Results and Discussion
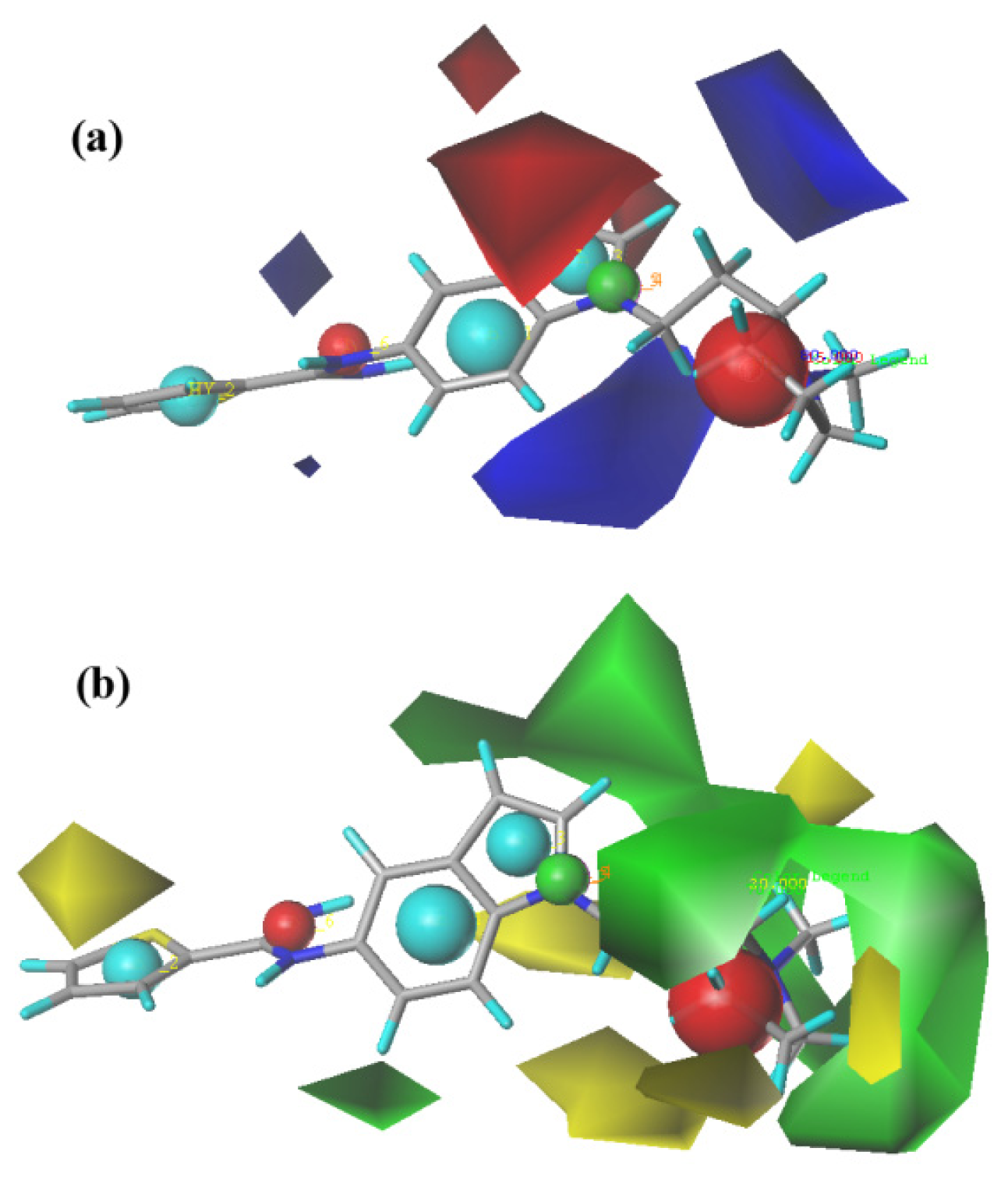
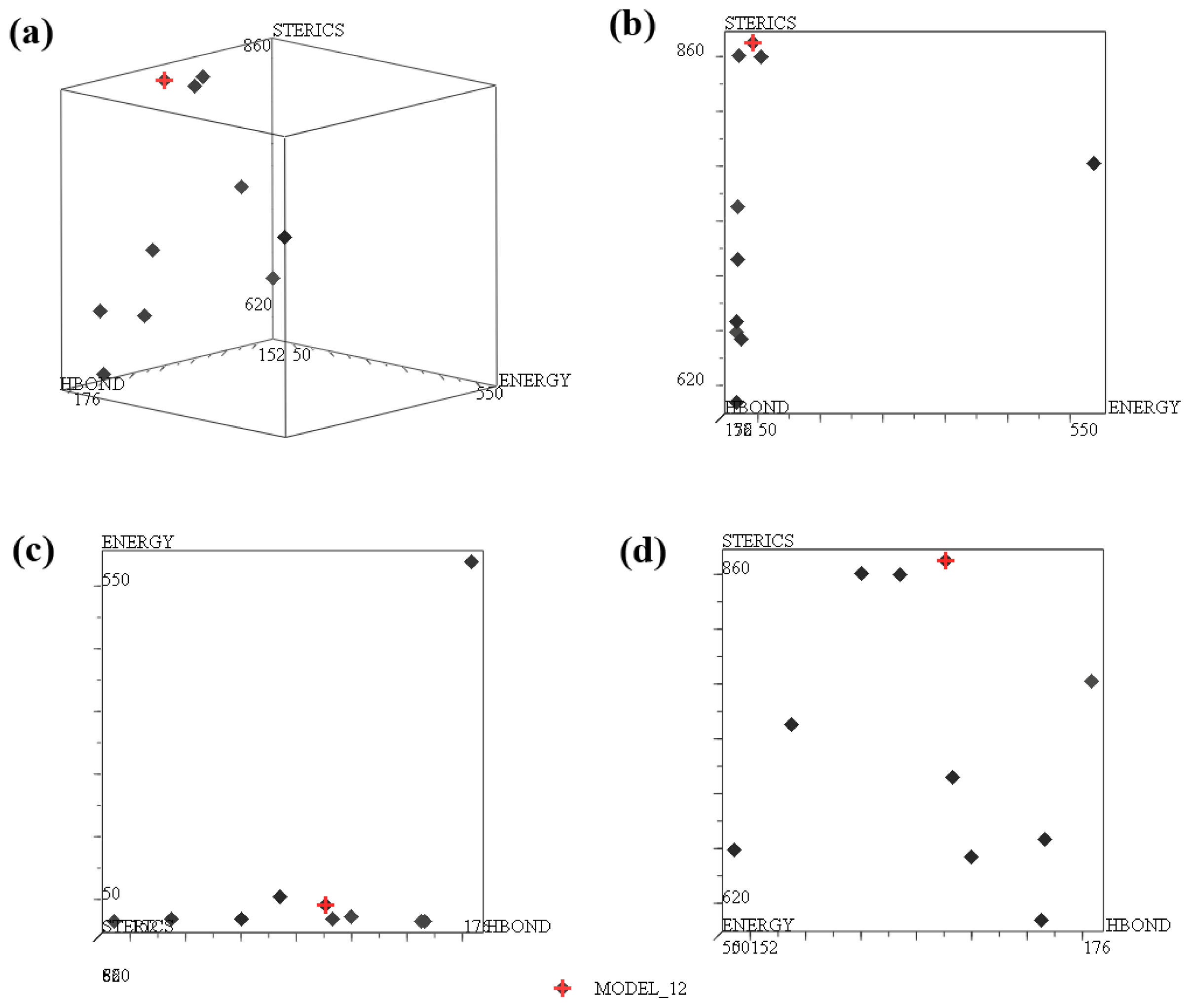
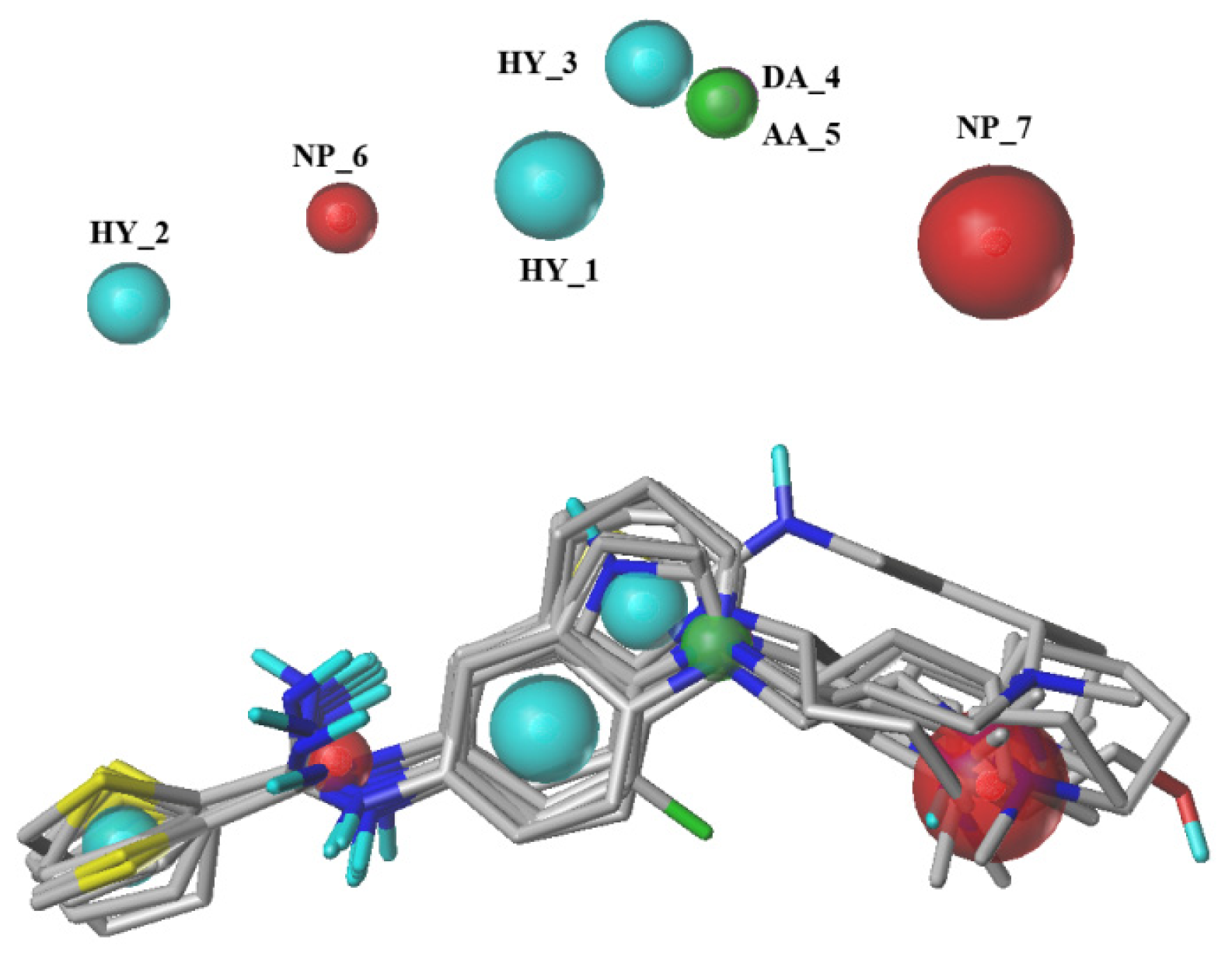
2.1. Pharmacophore Results
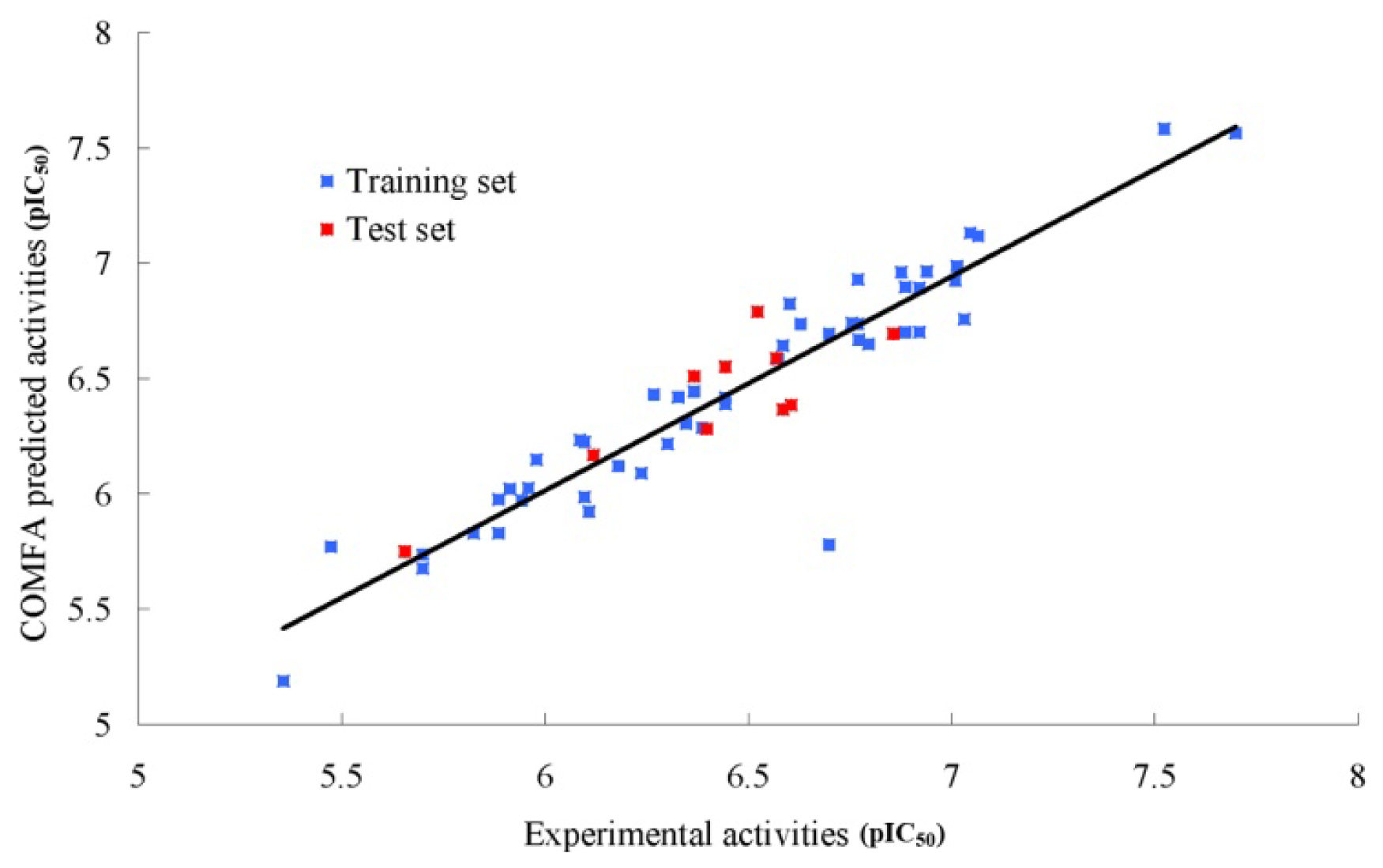
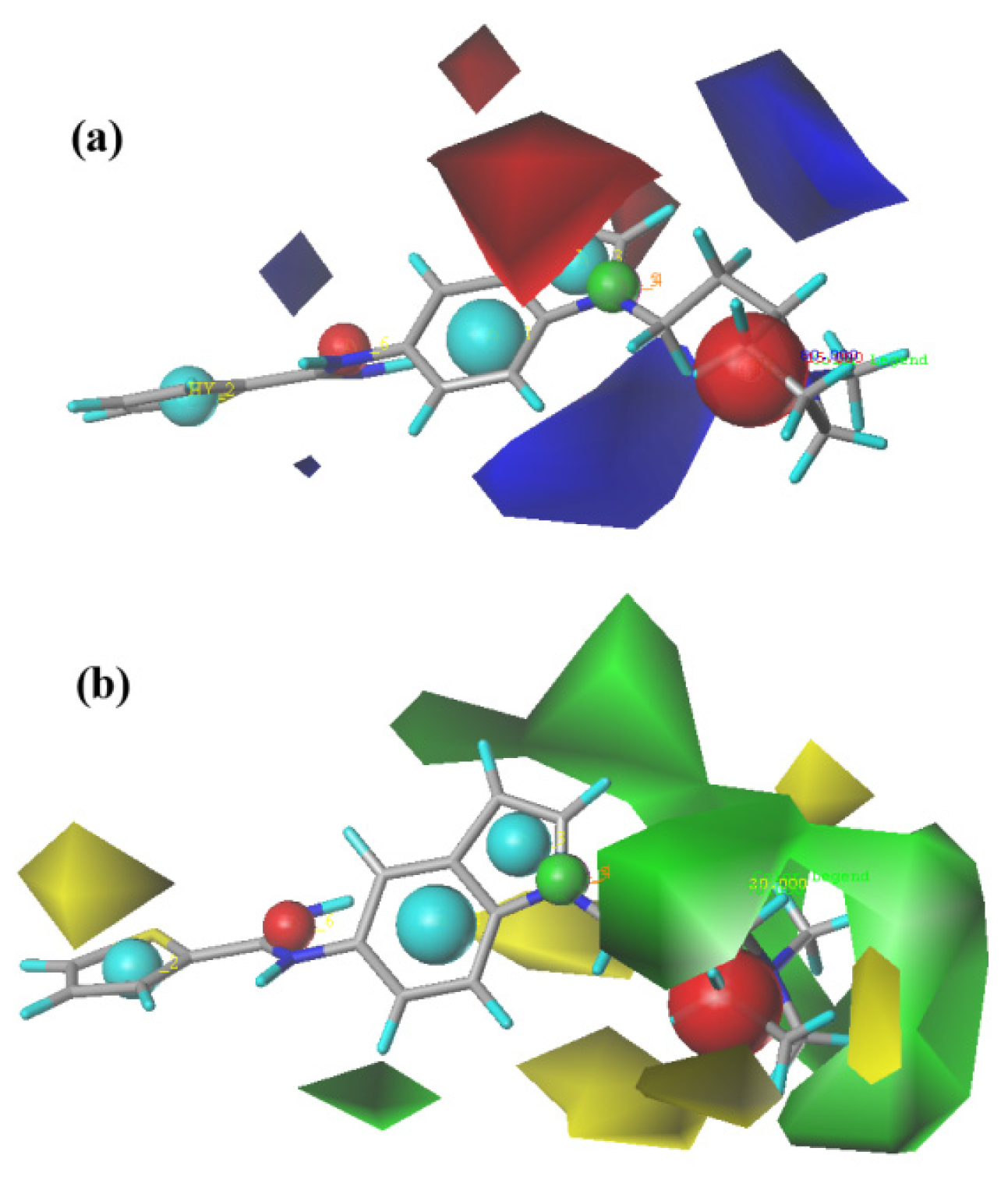
2.2. CoMFA (Comparative Molecular Field Analysis) Statistical Results
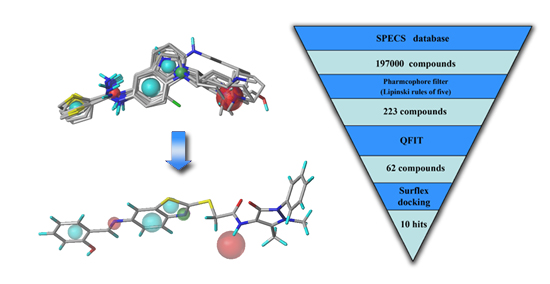
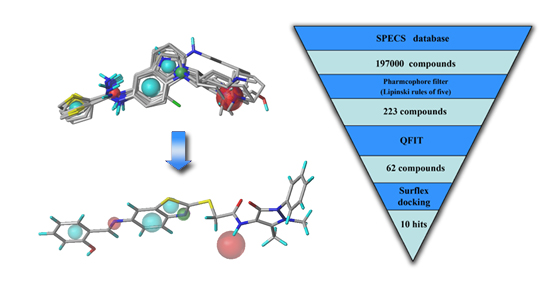
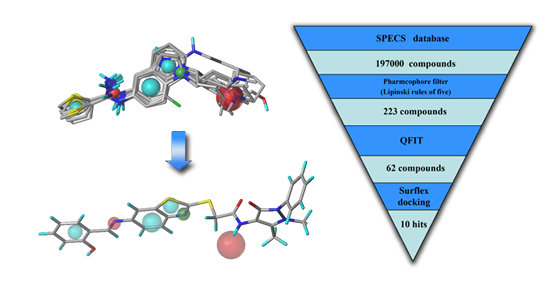
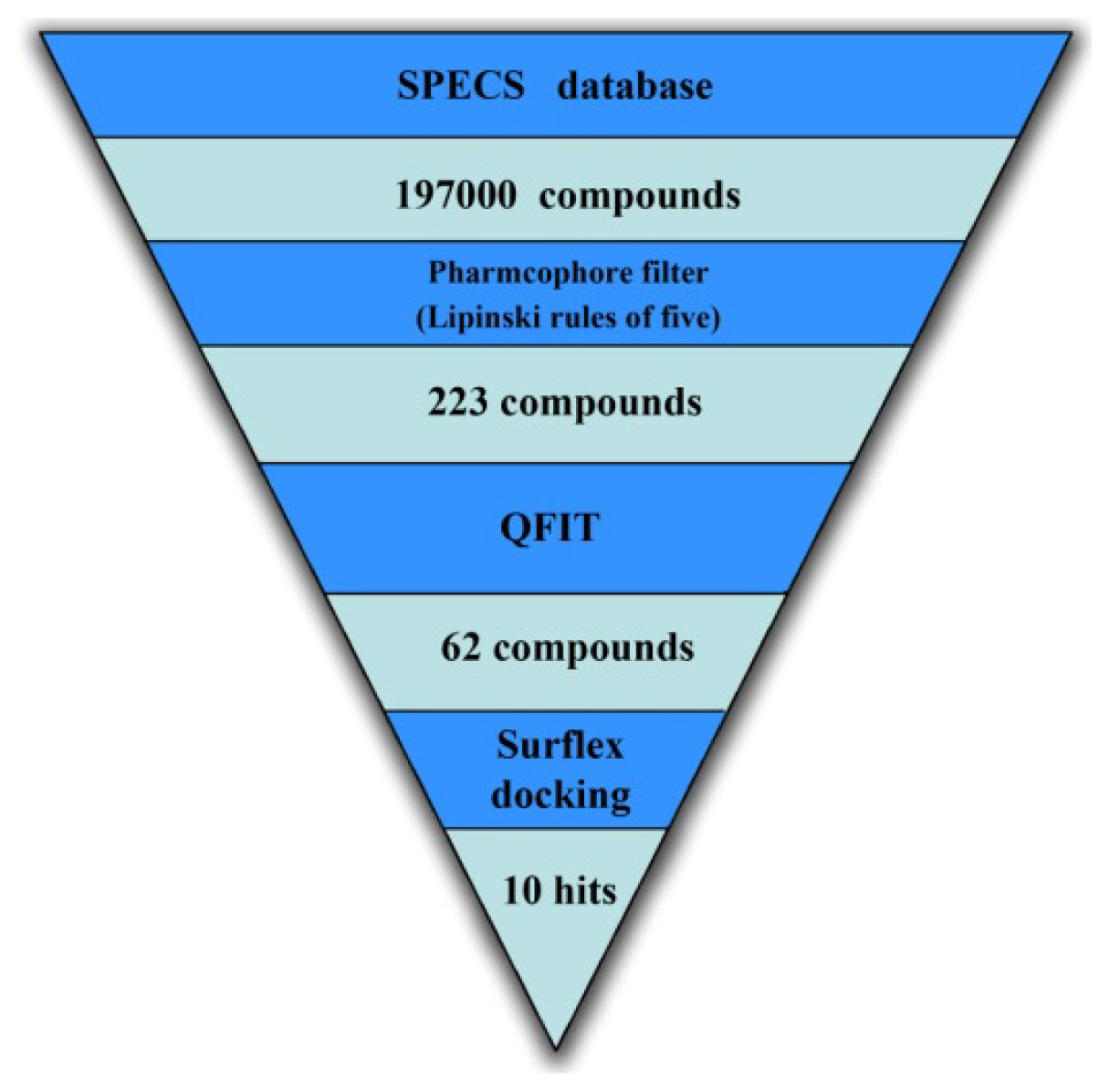
2.3. Virtual Screening
2.3.1. Database Searching
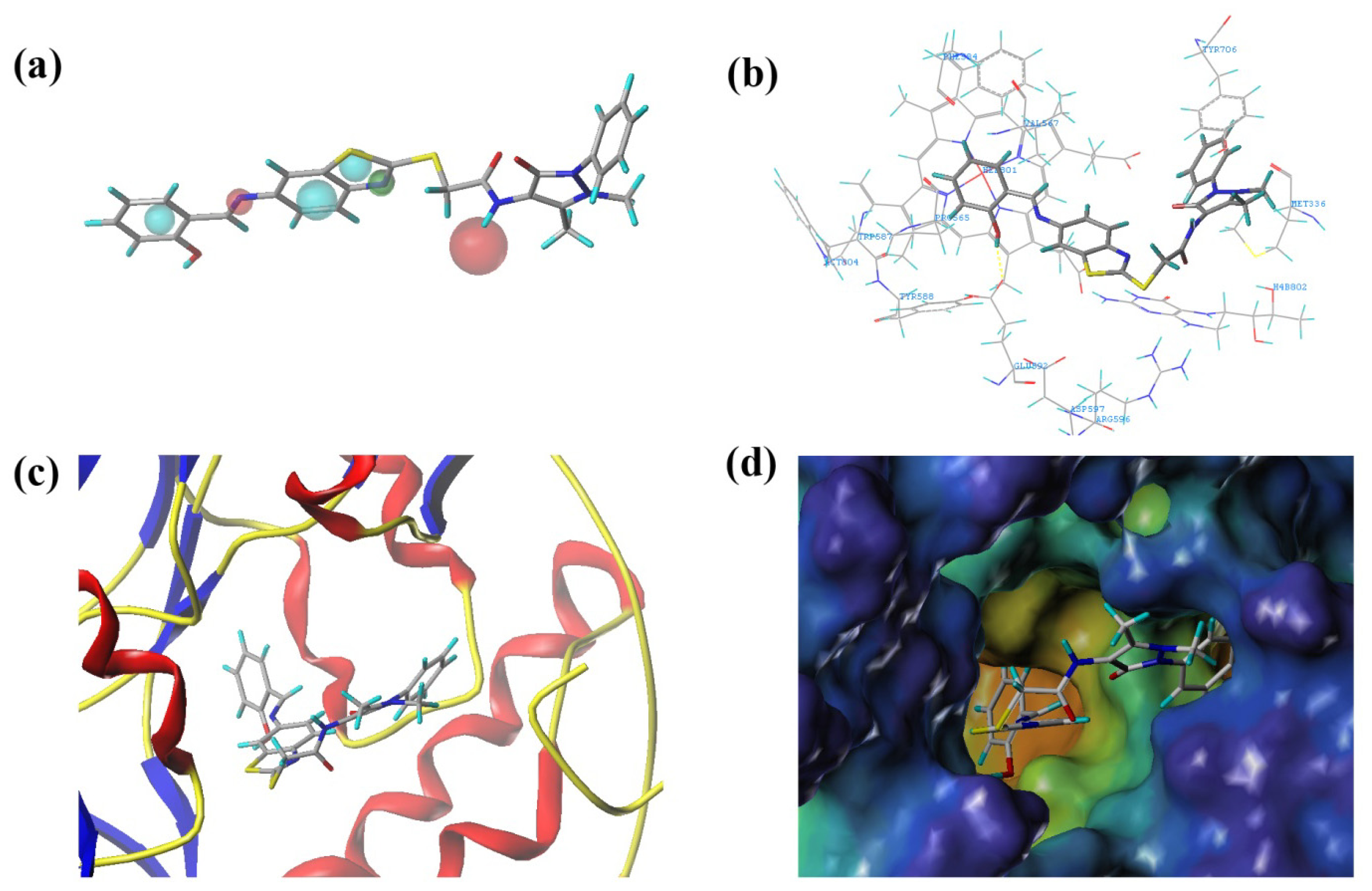
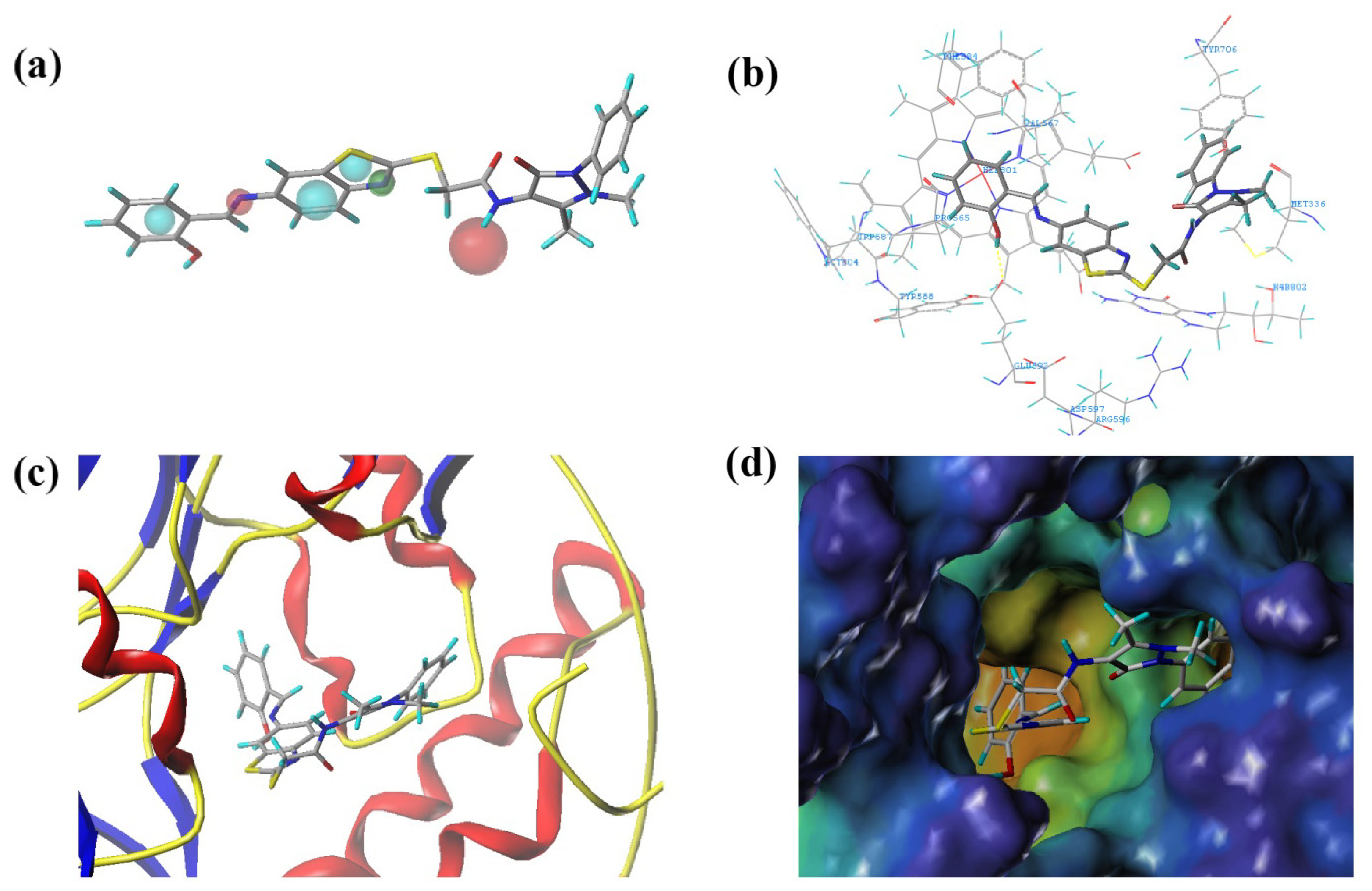
2.3.2. Molecular Docking
3. Experimental Section
3.1. Compounds and Biological Data
3.2. Pharmacophore Generation
3.3. CoMFA Field Calculation Partial Least Square Analysis
3.4. Virtual Screening
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsG.X. performed all experiments and data treatments. Writing was done by G.X., Y.C., K.S., X.W. and F.L., and management and submission tasks were done by F.L., Y.H. and G.X.
References
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J 2001, 357, 593–615. [Google Scholar]
- Stuehr, D.J. Structure-function aspects in the nitric oxide synthases. Annu. Rev. Pharmacol. Toxicol 1997, 37, 339–359. [Google Scholar]
- Gabriela, M.; Subhash, C.A.; Jailall, R.; Shawn, P.M.; Suman, R.; John, S.A.; Dongqin, Z.; Frank, P. First-in-class, dual-action, 3,5-disubstituted indole derivatives having human nitric oxide synthase (nNOS) and norepinephrine reuptake inhibitory (NERI) activity for the treatment of neuropathic pain. J. Med. Chem 2012, 55, 3488–3501. [Google Scholar]
- Mustafa, A.K.; Gadalla, M.M.; Snyder, S.H. Signaling by gasotransmitters. Sci. Signal 2009, 2. [Google Scholar] [CrossRef]
- Hao, J.X.; Xu, X.J. Treatment of a chronic allodynia-like response in spinally injured rats: Effects of systemically administered nitric oxide synthase inhibitors. Pain 1996, 66, 313–319. [Google Scholar]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacol. Rev 1991, 43, 109–142. [Google Scholar]
- Vallance, P.; Leiper, J. Blocking NO synthesis: How, where and why? Nat. Rev. Drug Discov 2002, 1, 939–950. [Google Scholar]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci 2007, 8, 766–775. [Google Scholar]
- Tanabe, M.; Nagatani, Y.; Saitoh, K.; Takasu, K.; Ono, H. Pharmacological assessments of nitric oxide synthase isoforms and downstream diversity of NO signaling in the maintenance of thermal and mechanical hypersensitivity after peripheral nerve injury in mice. Neuropharmacology 2009, 56, 702–708. [Google Scholar]
- Miclescu, A.; Gordh, T. Nitric oxide and pain: “Something old, something new”. Acta Anaesthesiol. Scand 2009, 53, 1107–1120. [Google Scholar]
- Gruber, H.J.; Bernecker, C.; Lechner, A.; Weiss, S.; Wallner-Blazek, M.; Meinitzer, A.; Höbarth, G.; Renner, W.; Fauler, G.; Horejsi, R.; et al. Increased nitric oxide stress is associated with migraine. Cephalalgia 2010, 30, 486–492. [Google Scholar]
- Payne, J.E.; Bonnefous, C.; Symons, K.T.; Nguyen, P.M.; Sablad, M.; Rozenkrants, N.; Zhang, Y.; Wang, L.; Yazdani, N.; Shiau, A.K.; et al. Discovery of dual inducible/neuronal nitric oxide synthase (iNOS/nNOS) inhibitor development candidate 4-((2-cyclobutyl-1H-imidazo[4,5-b]pyrazin-1-yl)methyl)-7,8-difluoroquinolin-2(1H)-one (KD7332) part 2: Identification of a novel, potent, and selective series of benzimidazole-quinolinone iNOS/nNOS dimerization inhibitors that are orally active in pain models. J. Med. Chem 2010, 53, 7739–7755. [Google Scholar]
- Erdal, E.P.; Litzinger, E.A.; Seo, J.; Zhu, Y.; Ji, H.; Silverman, R.B. Selective neuronal nitric oxide synthase inhibitors. Curr. Top. Med. Chem 2005, 5, 603–624. [Google Scholar]
- Silverman, R.B. Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc. Chem. Res 2009, 42, 439–451. [Google Scholar]
- Ramnauth, J.; Renton, P.; Dove, P.; Annedi, S.C.; Speed, J.; Silverman, S.; Mladenova, G.; Maddaford, S.P.; Zinghini, S.; Rakhit, S.; et al. 1,2,3,4-Tetrahydroquinoline-based selective human neuronal nitric oxide synthase (nNOS) inhibitors: Lead optimization studies resulting in the identification of N-(1-(2-(methylamino)ethyl)-1,2,3,4-tetrahydroquinolin-6-yl)thiophene-2-carboximi damide as a preclinical development candidate. J. Med. Chem 2012, 55, 2882–2893. [Google Scholar]
- Patman, J.; Bhardwaj, N.; Ramnauth, J.; Annedi, S.C.; Renton, P.; Maddaford, S.P.; Rakhit, S.; Andrews, J.S. Novel 2-aminobenzothiazoles as selective neuronal nitric oxide synthase inhibitors. Bioorg. Med. Chem. Lett 2007, 17, 2540–2544. [Google Scholar]
- Renton, P.; Speed, J.; Maddaford, S.; Annedi, S.C.; Ramnauth, J.; Rakhit, S.; Andrews, J. 1,5-Disubstituted indole derivatives as selective human neuronal nitric oxide synthase inhibitors. Bioorg. Med. Chem. Lett 2011, 21, 5301–5304. [Google Scholar]
- Maddaford, S.; Renton, P.; Speed, J.; Annedi, S.C.; Ramnauth, J.; Rakhit, S.; Andrews, J.; Mladenova, G.; Majuta, L.; Porreca, F. 1,6-Disubstituted indole derivatives as selective human neuronal nitric oxide synthase inhibitors. Bioorg. Med. Chem. Lett 2011, 21, 5234–5238. [Google Scholar]
- Annedi, S.C.; Maddaford, S.P.; Ramnauth, J.; Renton, P.; Speed, J.; Rakhit, S.; Andrews, J.S.; Porreca, F. 3,5-Disubstituted indole derivatives as selective human neuronal nitric oxide synthase (nNOS) inhibitors. Bioorg. Med. Chem. Lett 2012, 22, 1980–1984. [Google Scholar]
- Annedi, S.C.; Ramnauth, J.; Maddaford, S.P.; Renton, P.; Rakhit, S.; Mladenova, G.; Dove, P.; Silverman, S.; Andrews, J.S.; Felice, M.D.; et al. Discovery of cis-N-(1-(4-(methylamino) cyclohexyl)indolin-6-yl)thiophene-2-carboximidamide: a 1,6-disubstituted indoline derivative as a highly selective inhibitor of human neuronal nitric oxide synthase (nNOS) without any cardiovascular liabilities. J. Med. Chem 2012, 55, 943–955. [Google Scholar]
- Annedi, S.C.; Maddaford, S.P.; Mladenova, G.; Ramnauth, J.; Rakhit, S.; Andrews, J.S.; Lee, D.K.; Zhang, D.; Porreca, F.; Bunton, D.; et al. Discovery of N-(3-(1-methyl-1,2,3,6-tetrahydropyridin-4-yl)-1H-indol-6-yl) thiophene-2-carboximidamide as a selective inhibitor of human neuronal nitric oxide synthase (nNOS) for the treatment of pain. J. Med. Chem 2011, 54, 7408–7416. [Google Scholar]
- Ramnauth, J.; Speed, J.; Maddaford, S.P.; Dove, P.; Annedi, S.C.; Renton, P.; Rakhit, S.; Andrews, J.; Silverman, S.; Mladenova, G.; et al. Design, synthesis, and biological evaluation of 3,4-dihydroquinolin-2(1H)-one and 1,2,3,4-tetrahydroquinoline-based selective human neuronal nitric oxide synthase (nNOS) inhibitors. J. Med. Chem 2011, 54, 5562–5575. [Google Scholar]
- Xue, F.; Li, H.; Delker, S.L.; Fang, J.; Martásek, P.; Roman, L.J.; Poulos, T.L.; Silverman, R.B. Potent, highly selective, and orally bioavailable gem-difluorinated monocationic inhibitors of neuronal nitric oxide synthase. J. Am. Chem. Soc 2010, 132, 14229–14238. [Google Scholar]
- Huang, H.; Ji, H.; Li, H.; Jing, Q.; Labby, K.J.; Martásek, P.; Roman, L.J.; Poulos, T.L.; Silverman, R.B. Selective monocationic inhibitors of neuronal nitric oxide synthase. Binding mode insights from molecular dynamics simulations. J. Am. Chem. Soc 2012, 134, 11559–11572. [Google Scholar]
- Delker, S.L.; Ji, H.; Li, H.; Jamal, J.; Fang, J.; Xue, F.; Silverman, R.B.; Poulos, T.L. Unexpected binding modes of nitric oxide synthase inhibitors effective in the prevention of a cerebral palsy phenotype in an animal model. J. Am. Chem. Soc 2010, 132, 5437–5442. [Google Scholar]
- Klebe, G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discov. Today 2006, 11, 580–594. [Google Scholar]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc 1988, 110, 5959–5967. [Google Scholar]
- Xu, G.; Chu, Y.; Jiang, N.; Yang, J.; Li, F. The Three Dimensional Quantitative Structure Activity Relationships (3D-QSAR) and docking studies of curcumin derivatives as androgen receptor antagonists. Int. J. Mol. Sci 2012, 13, 6138–6155. [Google Scholar]
- SYBYL®-X Suite. Molecular Modeling from Sequence through Lead Optimization. Available online: http://www.certara.com/products/molmod/sybyl-x accessed on 12 May 2014.
- Zhao, X.; Yuan, M.; Huang, B.; Ji, H.; Zhu, L. Ligand-based pharmacophore model of N-Aryl and N-Heteroaryl piperazine alpha 1A-adrenoceptors antagonists using GALAHAD. J. Mol. Graph. Model 2010, 29, 126–136. [Google Scholar]
- Caballero, J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J. Mol. Graph. Model 2010, 29, 363–371. [Google Scholar]
- The Specs.net Chemistry. Available online: http://www.specs.net accessed on 12 May 2014.
- Shaikh, M.S.; Mittal, A.; Bharatam, P.V. Design of fructose-2,6-bisphosphatase inhibitors: A novel virtual screening approach. J. Mol. Graph. Model 2008, 26, 900–906. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 1997, 23, 3–25. [Google Scholar]
- Gugan, K.; Thirumurthy, M.; Changdev, G.G.; Seung, J.C. A combined 3D QSAR and pharmacophore-based virtual screening for the identification of potent p38 MAP kinase inhibitors: An in silico approach. Med. Chem. Res 2013, 22, 1773–1787. [Google Scholar]
- Lan, P.; Sun, J.R.; Chen, W.N.; Sun, P.H.; Chen, W.M. Molecular modelling studies on d-annulated benzazepinones as VEGF-R2 kinase inhibitors using docking and 3D-QSAR. J. Enzym. Inhib. Med. Chem 2011, 26, 367–377. [Google Scholar]
- Clark, M.; Cramer, R.D.; Opdenbosch, N.V. Validation of the general purpose tripos 5.2 force field. J. Comput. Chem 1989, 10, 982–1012. [Google Scholar]
- Lan, P.; Chen, W.N.; Xiao, G.K.; Sun, P.H.; Chen, W.M. 3D-QSAR and docking studies on pyrazolo[4,3-h]qinazoline-3-carboxamides as cyclin-dependent kinase 2 (CDK2) inhibitors. Bioorg. Med. Chem. Lett 2010, 20, 6764–6772. [Google Scholar]
- Pirhadi, S.; Ghasemi, J.B. 3D-QSAR analysis of human immunodeficiency virus entry-1 inhibitors by CoMFA and CoMSIA. Eur. J. Med. Chem 2010, 45, 4897–4903. [Google Scholar]
- Xu, G.; Zhou, Z.; Li, F. Combined 3D-QSAR modeling and molecular docking studies on naphthoquinone analogs as proteasome inhibitors. Lett. Drug Des. Discov 2013, 10, 129–144. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | SPECIFICITY | N_HITS | FEATS | PARETO | ENERGY | STERICS | HBOND |
|---|---|---|---|---|---|---|---|
| MOEDL_001 | 4.180 | 4 | 6 | 0 | 15.60 | 666.7 | 173.3 |
| MOEDL_002 | 3.881 | 8 | 7 | 0 | 15.44 | 703.1 | 161.8 |
| MOEDL_003 | 4.858 | 6 | 8 | 0 | 18.53 | 750.4 | 155.1 |
| MOEDL_004 | 4.108 | 6 | 6 | 0 | 18.62 | 712.0 | 166.6 |
| MOEDL_005 | 3.823 | 9 | 7 | 0 | 20.15 | 852.7 | 162.7 |
| MOEDL_006 | 3.735 | 6 | 7 | 0 | 17.54 | 714.7 | 160.5 |
| MOEDL_007 | 3.902 | 9 | 7 | 0 | 58.38 | 705.2 | 179.2 |
| MOEDL_008 | 4.051 | 6 | 6 | 0 | 19.3 | 784.6 | 171.4 |
| MOEDL_009 | 4.036 | 3 | 6 | 0 | 40.81 | 845.3 | 159.5 |
| MOEDL_010 | 3.393 | 5 | 9 | 0 | 35.12 | 612.2 | 178.2 |
| MOEDL_011 | 3.158 | 6 | 5 | 0 | 22.40 | 635.3 | 178.9 |
| MOEDL_012 b | 5.138 | 5 | 7 | 0 | 41.13 | 870.1 | 166.1 |
| MOEDL_013 | 4.048 | 6 | 6 | 0 | 17.75 | 732.8 | 157.0 |
| MOEDL_014 | 4.124 | 6 | 6 | 0 | 19.25 | 861.2 | 160.2 |
| MOEDL_015 | 3.867 | 8 | 7 | 0 | 19.99 | 567.7 | 175.3 |
| MOEDL_016 | 4.050 | 5 | 6 | 0 | 23.76 | 834.0 | 165.7 |
| MOEDL_017 | 4.053 | 5 | 6 | 0 | 14.97 | 658.9 | 150.8 |
| MOEDL_018 | 4.058 | 5 | 6 | 0 | 55.07 | 859.6 | 162.8 |
| MOEDL_019 | 5.128 | 4 | 7 | 0 | 23.06 | 654.1 | 168.0 |
| MOEDL_020 | 4.050 | 5 | 6 | 0 | 16.91 | 748.1 | 158.3 |
| No. | Structure | pIC50 | ||
|---|---|---|---|---|
| Observed | Predicted | |||
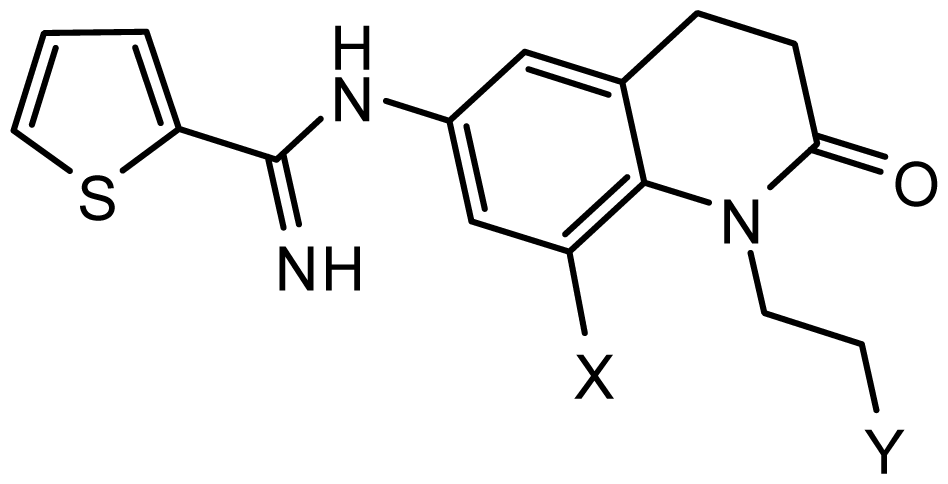
 series 1 [22] | ||||
| - | X | Y | - | - |
| 1 | H | N(CH3)2 | 6.237 | 6.089 |
| 2 * | H | N(Et)2 | 5.656 | 5.750 |
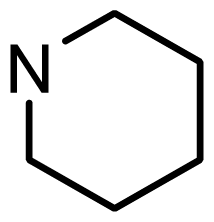
| 3 | H |  | 6.108 | 5.922 |
| 4 | H |  | 6.796 | 6.650 |
| 5 | H |  | 5.979 | 6.148 |
| 6 | F | N(CH3)2 | 5.474 | 5.770 |
| 7 | H | CH2N(CH3)2 | 5.943 | 5.971 |
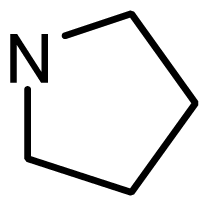
| 8 | H |  | 5.914 | 6.021 |
 series 2 [15,22] | ||||
| - | X | R | - | - |
| 9 * | H | −CH2CH2CH2N(CH3)2 | 6.569 | 6.588 |
| 10 | H | −CH2CH2NCH3 | 6.754 | 6.741 |
| 11 * | H | −CH2CH2N CH2CH3 | 6.857 | 6.694 |
| 12 | H | −CH2CH2NCH(CH3)2 | 6.573 | 6.585 |
| 13 | H | −CH2CH2N(CH3) (C2H5) | 7.013 | 6.987 |
| 14 * | H | −CH2CH2N(CH3)2 | 6.367 | 6.510 |
| 15 | H | −CH2CH2N(C2H5)2 | 6.585 | 6.642 |
| 16 | F | −CH2CH2N(C2H5)2 | 7.032 | 6.757 |
| 17 | H | − (CH2)3NCH3 | 6.629 | 6.736 |
| 18 | H | −CH2CH2N(CH3) (CH2)2OH | 6.876 | 6.960 |
| 19 | H | − (CH2)2NH(CH2)2OH | 6.939 | 6.964 |
| 20 | H | − (CH2)3NH(CH2)2OH | 6.772 | 6.667 |
| 21 | H |  | 7.009 | 6.925 |
| 22 | H |  | 6.886 | 6.896 |
| 23 * | H | 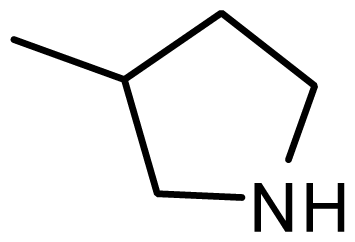 | 6.606 | 6.385 |
| 24 | H | 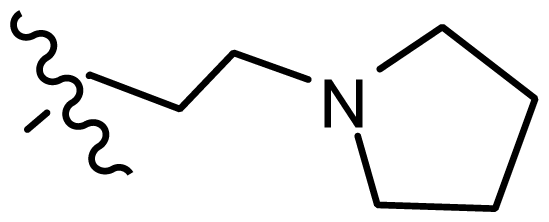 | 7.066 | 7.118 |
| 25 | H |  | 6.086 | 6.233 |
| 26 | H |  | 6.268 | 6.430 |
| 27 * | H |  | 6.444 | 6.550 |
 series 3 [21] | ||||
| - | X | R | - | - |
| 28 | S |  | 6.699 | 6.694 |
| 29 | S |  | 6.097 | 6.225 |
| 30 | S |  | 6.921 | 6.701 |
| 31 | S |  | 5.824 | 5.830 |
| 32 | S |  | 6.347 | 6.304 |
 series 4 [17] | ||||
| - | X | R | - | - |
| 33 | S | N-(1-(3-(dimethylamino)propyl)- | 6.328 | 6.419 |
| 34 * | S | N-(1-(3-(cyclopropylamino)propyl)- | 6.585 | 6.366 |
| 35 | S | N-(1-(3-morpholinopropyl)- | 6.181 | 6.120 |
| 36 | S | N-(1-(3-((1-ethylpyrrolidin-2-yl)methylamino)propyl)- | 6.886 | 6.700 |
| 37 | S | N-(1-(3-adamantanaminopropyl)- | 6.444 | 6.388 |
| 38 | S | N-(1-(2-(dimethylamino)ethyl)- | 6.770 | 6.736 |
| 39 | S | N-(1-(2-(piperidin-1-yl)ethyl)- | 6.770 | 6.930 |
| 40 | S | N-(1-(2-(1-methylpiperidin-2-yl)ethyl)- | 7.046 | 7.131 |
| 41 | S | (S) N-(1-(2-(1-methylpyrrolidin-2-yl)ethyl)- | 7.700 | 7.564 |
| 42 | O | N-(1-(2-(1-methylpyrrolidin-2-yl)ethyl)- | 6.602 | 6.824 |
| 43 | S | N-(1-(1-methylazepan-4-yl)- | 6.921 | 6.893 |
| 44 | O | N-(1-(1-methylazepan-4-yl)- | 6.367 | 6.443 |
| 45 * | S | N-(1-(8-methyl-8-azabicyclo[3.2.1]octan-3-yl)- | 6.120 | 6.168 |
| 46 | S | N-(1-(quinuclidin-3-yl)- | 6.444 | 6.417 |
| 47 | S | N-(1-(1-methylpiperidin-4-yl)- | 6.387 | 6.286 |
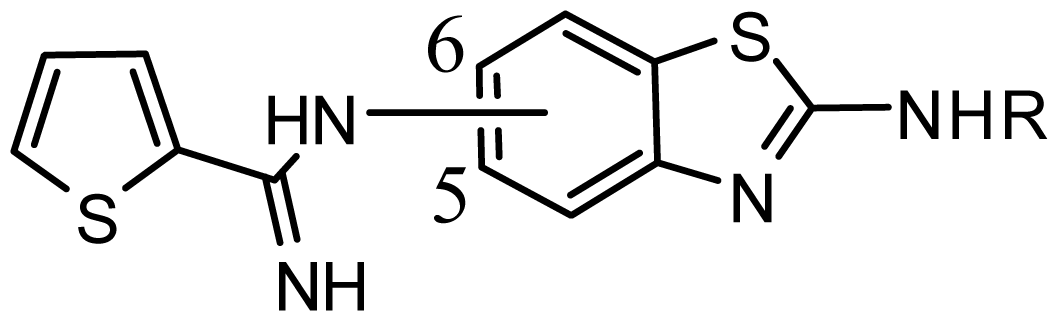 series 5 [16] | ||||
| Substituted | R | |||
| 48 | 5 | 2-(Pyridin-2-yl)ethyl | 5.959 | 6.025 |
| 49 | 5 | 2-Morpholinoethyl | 5.886 | 5.976 |
| 50 * | 5 | 1-Benzylpiperidin-4-yl | 6.398 | 6.281 |
| 51 | 5 | 1-(4-Fluorobenzyl)piperidin-4-yl | 6.097 | 5.986 |
| 52 | 5 | (±)-2-(1-Methylpyrrolidin-2-yl)ethyl | 7.523 | 7.582 |
| 53 | 6 | 2-(Pyridin-2-yl)ethyl | 5.886 | 5.83 |
| 54 | 6 | 2-Morpholinoethyl | 5.699 | 5.676 |
| 55 | 6 | 1-Benzylpiperidin-4-yl | 6.301 | 6.216 |
| 56 | 6 | 1-(4-Fluorobenzyl)piperidin-4-yl | 6.699 | 5.779 |
| 57 * | 6 | 2-(1H-Imidazol-5-yl)ethyl | 6.523 | 6.789 |
| 58 | 6 | 4-Bromophenethyl | 5.357 | 5.188 |
| 59 | 6 | Tetrahydro-2H-pyran-4-yl | 5.699 | 5.736 |
| SPECS ID | Structure | Dock Scores | QFIT |
|---|---|---|---|
| AG_205/36953325 |  | 8.29 | 65.74 |
| AG_205/11218159 | 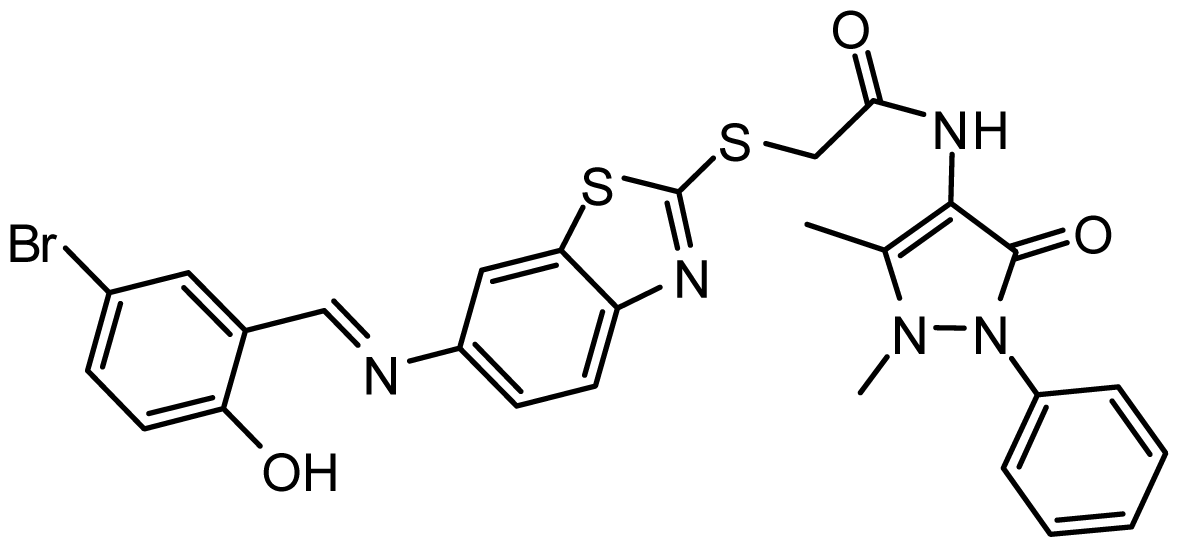 | 8.20 | 65.74 |
| AG_205/11218337 |  | 8.01 | 65.82 |
| AG_205/11218321 |  | 7.86 | 65.82 |
| AG_205/36564022 | 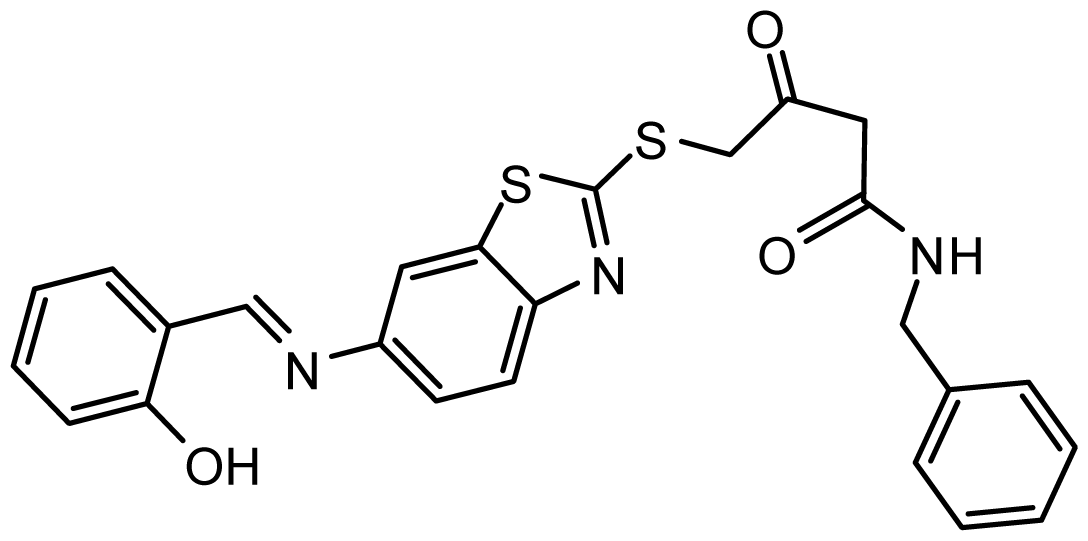 | 7.65 | 65.82 |
| AG_205/36953138 |  | 7.63 | 65.81 |
| AG_205/09949027 |  | 7.34 | 65.82 |
| AG_205/36953406 | 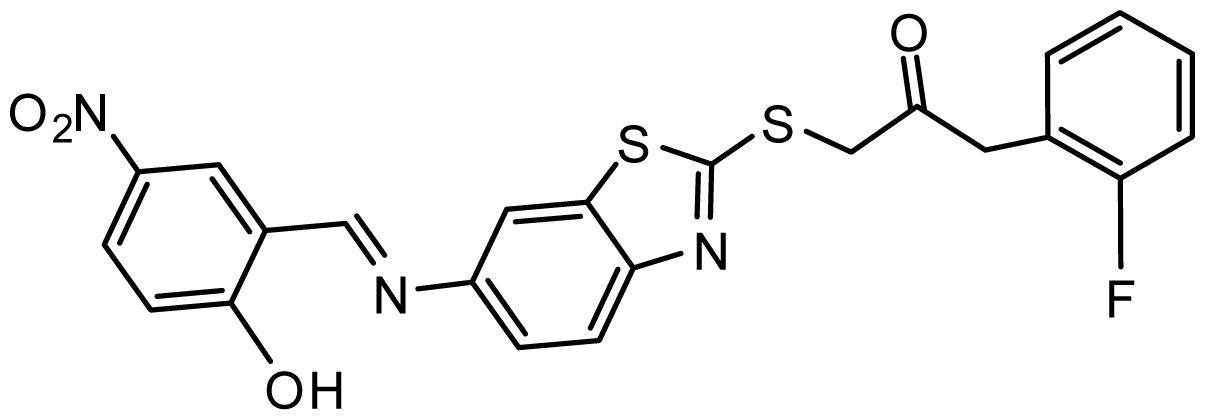 | 6.78 | 65.82 |
| AG_205/36265063 | 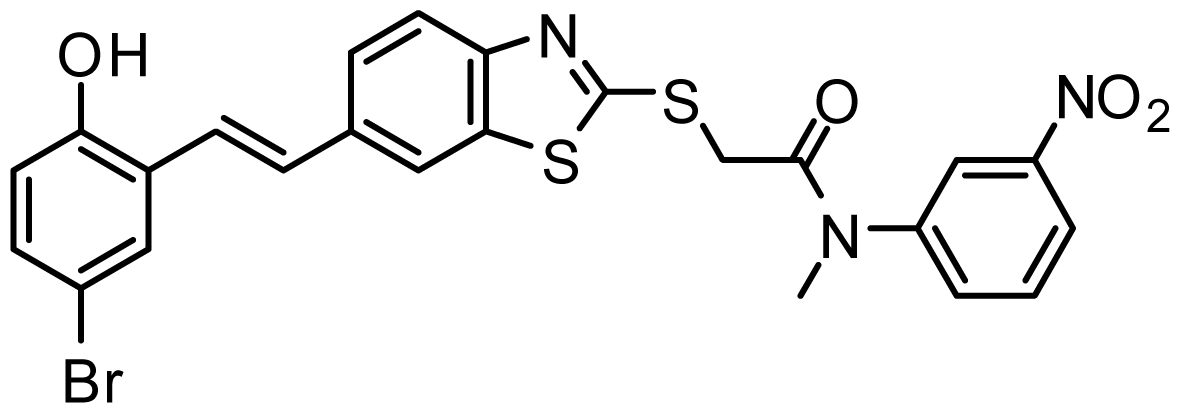 | 6.51 | 65.82 |
| AG_205/36940042 |  | 6.22 | 65.81 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, G.; Chen, Y.; Shen, K.; Wang, X.; Li, F.; He, Y. The Discovery of Potentially Selective Human Neuronal Nitric Oxide Synthase (nNOS) Inhibitors: A Combination of Pharmacophore Modelling, CoMFA, Virtual Screening and Molecular Docking Studies. Int. J. Mol. Sci. 2014, 15, 8553-8569. https://doi.org/10.3390/ijms15058553
Xu G, Chen Y, Shen K, Wang X, Li F, He Y. The Discovery of Potentially Selective Human Neuronal Nitric Oxide Synthase (nNOS) Inhibitors: A Combination of Pharmacophore Modelling, CoMFA, Virtual Screening and Molecular Docking Studies. International Journal of Molecular Sciences. 2014; 15(5):8553-8569. https://doi.org/10.3390/ijms15058553
Chicago/Turabian StyleXu, Guanhong, Yue Chen, Kun Shen, Xiuzhen Wang, Fei Li, and Yan He. 2014. "The Discovery of Potentially Selective Human Neuronal Nitric Oxide Synthase (nNOS) Inhibitors: A Combination of Pharmacophore Modelling, CoMFA, Virtual Screening and Molecular Docking Studies" International Journal of Molecular Sciences 15, no. 5: 8553-8569. https://doi.org/10.3390/ijms15058553