Colonization and Infection of the Skin by S. aureus: Immune System Evasion and the Response to Cationic Antimicrobial Peptides

Abstract
:1. Introduction
2. Host-Pathogen Interactions during S. aureus Skin Colonization and Infection
3. Methicillin-Resistant S. aureus (MRSA) Infection
4. Human AMPs Effective against S. aureus
4.1. Defensins
4.2. Cathelicidins
4.3. RNase7
4.4. Dermcidin
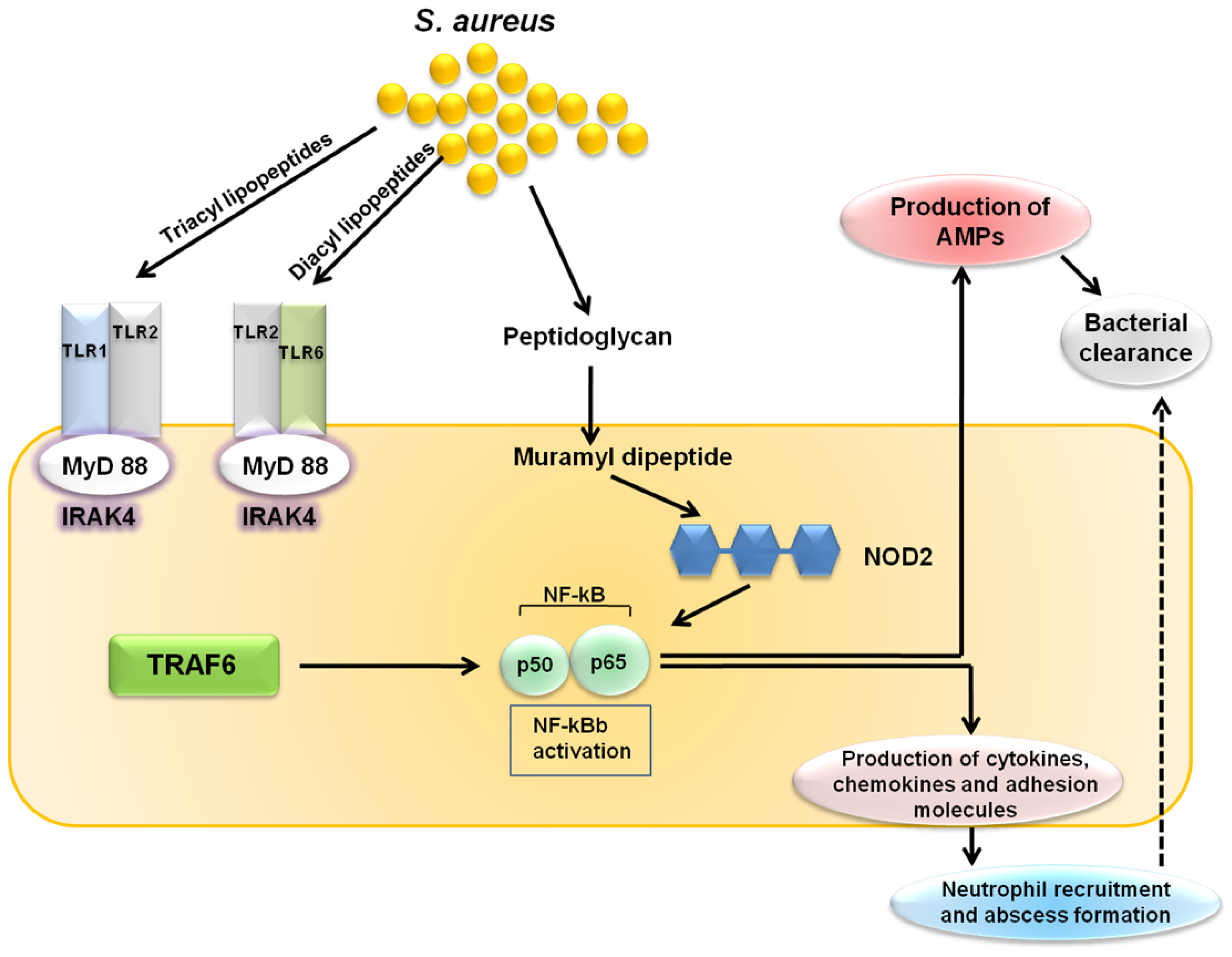
5. Cutaneous Host Defense Involving TLR-Mediated AMP Activity against S. aureus
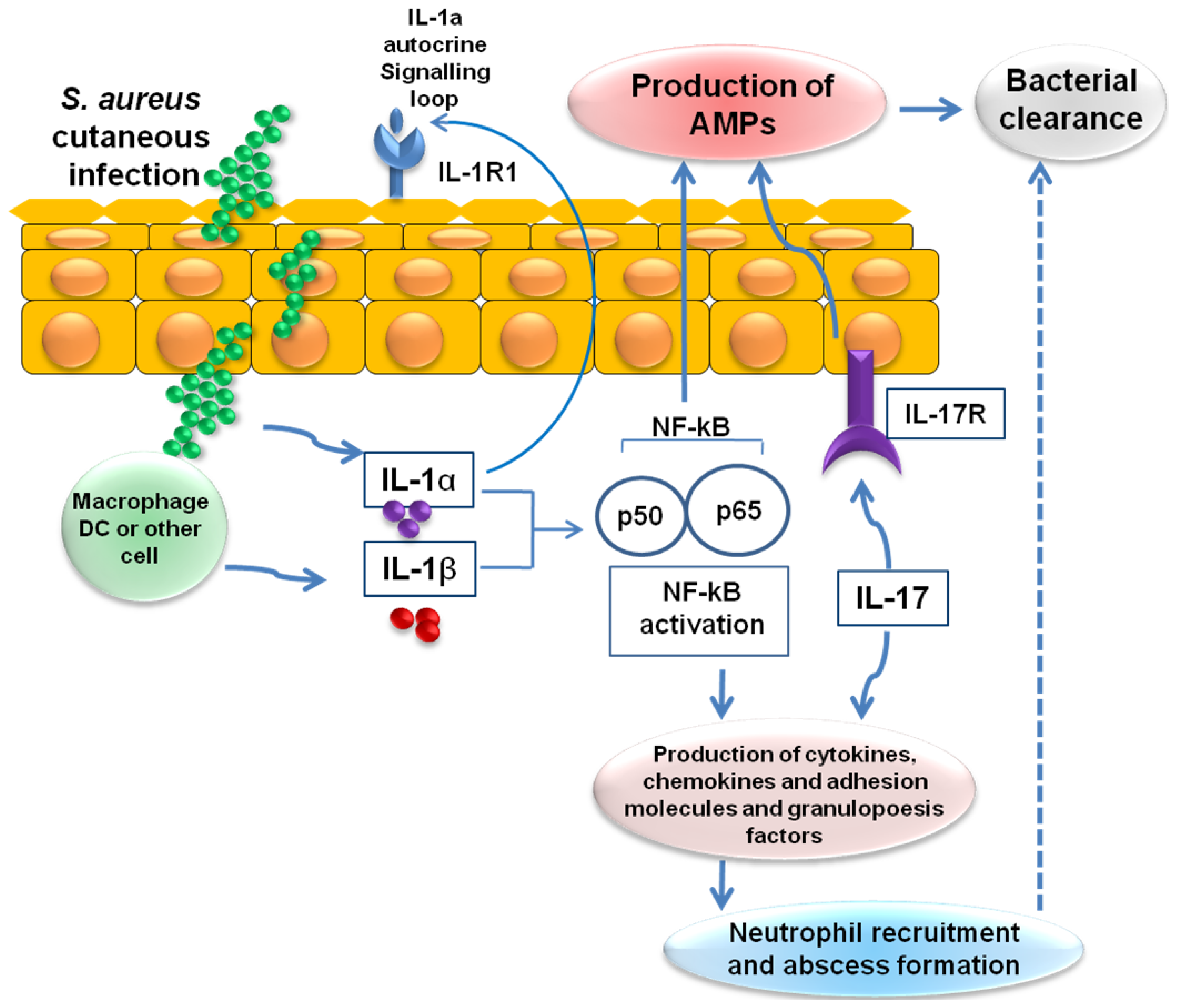
6. Cutaneous Host Defense Involving IL-1- and IL-17-Mediated AMP Activity against S. aureus
7. Mechanisms by Which S. aureus Evades Skin-Derived CAMPs
7.1. Secretion of Extracellular CAMP-Binding Molecules
7.2. Proteolytic Degradation of CAMPs by Secreted Proteases
7.3. Resistance to CAMPs through Reduction of Bacterial Surface Net Negative Charges
7.3.1. Modification of Phospholipids with l l -lysine
7.3.2. Modification of Teichoic Acids with d-alanine
7.4. Resistance to CAMPs through Alteration of Bacterial Cell Surface Hydrophobicity
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chambers, H.F. The changing epidemiology of Staphylococcus aureus? Emerg. Infect. Dis 2001, 7, 178–182. [Google Scholar]
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med 1998, 339, 520–532. [Google Scholar]
- Graham, P.L., 3rd; Lin, S. X.; Larson, E.L. A U.S. population-based survey of Staphylococcus aureus colonization. Ann. Intern. Med 2006, 144, 318–325. [Google Scholar]
- Kuehnert, M.J.; Kruszon-Moran, D.; Hill, H.A.; McQuillan, G.; McAllister, S.K.; Fosheim, G.; McDougal, L.K.; Chaitram, J.; Jensen, B.; Fridkin, S.K.; et al. Prevalence of Staphylococcus aureus nasal colonization in the United States, 2001–2002. J. Infect. Dis 2006, 193, 172–179. [Google Scholar]
- Perl, T.M.; Cullen, J.J.; Wenzel, R.P.; Zimmerman, M.B.; Pfaller, M.A.; Sheppard, D.; Twombley, J.; French, P.P.; Herwaldt, L.A.; et al. Intranasal mupirocin to prevent postoperative Staphylococcus aureus infections. N. Engl. J. Med 2002, 346, 1871–1877. [Google Scholar]
- Shopsin, B.; Mathema, B.; Martinez, J.; Ha, E.; Campo, M.L.; Fierman, A.; Krasinski, K.; Kornblum, J.; Alcabes, P.; Waddington, M.; Riehman, M.; et al. Prevalence of methicillin-resistant and methicillin-susceptible Staphylococcus aureus in the community. J. Infect. Dis 2000, 182, 359–362. [Google Scholar]
- Von Eiff, C.; Becker, K.; Machka, K.; Stammer, H.; Peters, G. Nasal carriage as a source of Staphylococcus aureus bacteremia. N. Engl. J. Med 2001, 344, 11–16. [Google Scholar]
- Herwaldt, L.A. Staphylococcus aureus nasal carriage and surgical-site infections. Surgery 2003, 134, S2–S9. [Google Scholar]
- Lin, Y.C.; Lauderdale, T.L.; Lin, H.M.; Chen, P.C.; Cheng, M.F.; Hsieh, K.S.; Liu, Y.C. An outbreak of methicillin-resistant Staphylococcus aureus infection in patients of a pediatric intensive care unit and high carriage rate among health care workers. J. Microbiol. Immunol. Infect 2007, 40, 325–334. [Google Scholar]
- Elston, D.M. Community-acquired methicillin-resistant Staphylococcus aureus. J. Am. Acad. Dermatol 2007, 56, 1–16. [Google Scholar]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol 2005, 3, 948–958. [Google Scholar]
- Kraus, D.; Peschel, A. Molecular mechanisms of bacterial resistance to antimicrobial peptides. Curr. Top. Microbiol. Immunol 2006, 306, 231–250. [Google Scholar]
- Glaser, R.; Becker, K.; von Eiff, C.; Meyer-Hoffert, U.; Harder, J. Decreased susceptibility of Staphylococcus aureus small colony variants (SCVs) towards human antimicrobial peptides. J. Invest. Dermatol 2014. [Google Scholar] [CrossRef]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar]
- Woolhouse, M.E.; Webster, J.P.; Domingo, E.; Charlesworth, B.; Levin, B.R. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat. Genet 2002, 32, 569–577. [Google Scholar]
- Gorwitz, R.J.; Kruszon-Moran, D.; McAllister, S.K.; McQuillan, G.; McDougal, L.K.; Fosheim, G.E.; Jensen, B.J.; Killgore, G.; Tenover, F.C.; Kuehnert, M.J. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001–2004. J. Infect. Dis 2008, 197, 1226–1234. [Google Scholar]
- Miller, L.G.; Diep, B.A. Clinical practice: Colonization, fomites, and virulence: Rethinking the pathogenesis of community-associated methicillin-resistant Staphylococcus aureus infection. Clin. Infect. Dis 2008, 46, 752–760. [Google Scholar]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; Program, N.C.S.; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and temporal diversity of the human skin microbiome. Science 2009, 324, 1190–1192. [Google Scholar]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol 2011, 9, 244–253. [Google Scholar]
- Miajlovic, H.; Fallon, P.G.; Irvine, A.D.; Foster, T.J. Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J. Allergy. Clin. Immunol 2010, 126, 1184–1190. [Google Scholar]
- Iwase, T.; Uehara, Y.; Shinji, H.; Tajima, A.; Seo, H.; Takada, K.; Agata, T.; Mizunoe, Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 2010, 465, 346–349. [Google Scholar]
- Cogen, A.L.; Yamasaki, K.; Sanchez, K.M.; Dorschner, R.A.; Lai, Y.; MacLeod, D.T.; Torpey, J.W.; Otto, M.; Nizet, V.; Kim, J.E.; et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J. Invest. Dermatol 2010, 130, 192–200. [Google Scholar]
- Lai, Y.; Cogen, A.L.; Radek, K.A.; Park, H.J.; Macleod, D.T.; Leichtle, A.; Ryan, A.F.; Di Nardo, A.; Gallo, R.L. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J. Invest. Dermatol 2010, 130, 2211–2221. [Google Scholar]
- Wanke, I.; Steffen, H.; Christ, C.; Krismer, B.; Gotz, F.; Peschel, A.; Schaller, M.; Schittek, B. Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J. Invest. Dermatol 2011, 131, 382–390. [Google Scholar]
- Braff, M.H.; Zaiou, M.; Fierer, J.; Nizet, V.; Gallo, R.L. Keratinocyte production of cathelicidin provides direct activity against bacterial skin pathogens. Infect. Immun 2005, 73, 6771–6781. [Google Scholar]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. A peptide antibiotic from human skin. Nature 1997, 387, 861. [Google Scholar]
- Kisich, K.O.; Howell, M.D.; Boguniewicz, M.; Heizer, H.R.; Watson, N.U.; Leung, D.Y. The constitutive capacity of human keratinocytes to kill Staphylococcus aureus is dependent on β-defensin 3. J. Invest. Dermatol 2007, 127, 2368–2380. [Google Scholar]
- Simanski, M.; Dressel, S.; Glaser, R.; Harder, J. RNase 7 protects healthy skin from Staphylococcus aureus colonization. J. Invest. Dermatol 2010, 130, 2836–2838. [Google Scholar]
- Ong, P.Y.; Ohtake, T.; Brandt, C.; Strickland, I.; Boguniewicz, M.; Ganz, T.; Gallo, R.L.; Leung, D.Y. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med 2002, 347, 1151–1160. [Google Scholar]
- Burian, M.; Rautenberg, M.; Kohler, T.; Fritz, M.; Krismer, B.; Unger, C.; Hoffmann, W.H.; Peschel, A.; Wolz, C.; Goerke, C. Temporal expression of adhesion factors and activity of global regulators during establishment of Staphylococcus aureus nasal colonization. J. Infect. Dis 2010, 201, 1414–1421. [Google Scholar]
- Cho, S.H.; Strickland, I.; Tomkinson, A.; Fehringer, A.P.; Gelfand, E.W.; Leung, D.Y. Preferential binding of Staphylococcus aureus to skin sites of Th2-mediated inflammation in a murine model. J. Invest. Dermatol 2001, 116, 658–663. [Google Scholar]
- Clarke, S.R.; Brummell, K.J.; Horsburgh, M.J.; McDowell, P.W.; Mohamad, S.A.; Stapleton, M.R.; Acevedo, J.; Read, R.C.; Day, N.P.; Peacock, S.J.; et al. Identification of in vivo-expressed antigens of Staphylococcus aureus and their use in vaccinations for protection against nasal carriage. J. Infect. Dis 2006, 193, 1098–1108. [Google Scholar]
- Weidenmaier, C.; Kokai-Kun, J.F.; Kristian, S.A.; Chanturiya, T.; Kalbacher, H.; Gross, M.; Nicholson, G.; Neumeister, B.; Mond, J.J.; Peschel, A. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat. Med 2004, 10, 243–245. [Google Scholar]
- Laouini, D.; Kawamoto, S.; Yalcindag, A.; Bryce, P.; Mizoguchi, E.; Oettgen, H.; Geha, R.S. Epicutaneous sensitization with superantigen induces allergic skin inflammation. J. Allergy. Clin. Immunol 2003, 112, 981–987. [Google Scholar]
- Clarke, S.R.; Mohamed, R.; Bian, L.; Routh, A.F.; Kokai-Kun, J.F.; Mond, J.J.; Tarkowski, A.; Foster, S.J. The Staphylococcus aureus surface protein IsdA mediates resistance to innate defenses of human skin. Cell Host Microbe 2007, 1, 199–212. [Google Scholar]
- Sieprawska-Lupa, M.; Mydel, P.; Krawczyk, K.; Wojcik, K.; Puklo, M.; Lupa, B.; Suder, P.; Silberring, J.; Reed, M.; Pohl, J.; et al. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother 2004, 48, 4673–4679. [Google Scholar]
- Zecconi, A.; Scali, F. Staphylococcus aureus virulence factors in evasion from innate immune defenses in human and animal diseases. Immunol. Lett 2013, 150, 12–22. [Google Scholar]
- Hallander, H.O.; Laurell, G.; Lofstrom, G. Enhancement of staphylococcal pathogenicity in the presence of penicillin. Acta Pathol. Microbiol. Scand 1966, 68, 463–464. [Google Scholar]
- Ohlsen, K.; Ziebuhr, W.; Koller, K.P.; Hell, W.; Wichelhaus, T.A.; Hacker, J. Effects of subinhibitory concentrations of antibiotics on α-toxin (hla) gene expression of methicillin-sensitive and methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother 1998, 42, 2817–2823. [Google Scholar]
- Shibl, A.M. Effect of antibiotics on production of enzymes and toxins by microorganisms. Rev. Infect. Dis 1983, 5, 865–875. [Google Scholar]
- Bernardo, K.; Pakulat, N.; Fleer, S.; Schnaith, A.; Utermohlen, O.; Krut, O.; Muller, S.; Kronke, M. Subinhibitory concentrations of linezolid reduce Staphylococcus aureus virulence factor expression. Antimicrob. Agents Chemother 2004, 48, 546–555. [Google Scholar]
- Schlievert, P.M.; Kelly, J.A. Clindamycin-induced suppression of toxic-shock syndrome— Associated exotoxin production. J. Infect. Dis 1984, 149, 471. [Google Scholar]
- Shibl, A.M. Role of Staphylococcus aureus exfoliatin toxin in staphylococcal infections in mice. Chemotherapy 1981, 27, 224–227. [Google Scholar]
- Veringa, E. M.; Verhoef, J. Influence of subinhibitory concentrations of clindamycin on opsonophagocytosis of Staphylococcus aureus, a protein-A-dependent process. Antimicrob. Agents Chemother 1986, 30, 796–797. [Google Scholar]
- Beilman, G.J.; Sandifer, G.; Skarda, D.; Jensen, B.; McAllister, S.; Killgore, G.; Srinivasan, A. Emerging infections with community-associated methicillin-resistant Staphylococcus aureus in outpatients at an Army Community Hospital. Surg. Infect. (Larchmt) 2005, 6, 87–92. [Google Scholar]
- Daum, R.S. Clinical practice: Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N. Engl. J. Med 2007, 357, 380–390. [Google Scholar]
- David, M.Z.; Daum, R.S. Community-associated methicillin-resistant Staphylococcus aureus: Epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev 2010, 23, 616–687. [Google Scholar]
- DeLeo, F.R.; Otto, M.; Kreiswirth, B.N.; Chambers, H.F. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010, 375, 1557–1568. [Google Scholar]
- Deurenberg, R.H.; Stobberingh, E.E. The evolution of Staphylococcus aureus. Infect. Genet. Evol 2008, 8, 747–763. [Google Scholar]
- Mediavilla, J.R.; Chen, L.; Mathema, B.; Kreiswirth, B.N. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA). Curr. Opin. Microbiol 2012, 15, 588–595. [Google Scholar]
- Chavez, T.T.; Decker, C.F. Health care-associated MRSA versus community-associated MRSA. Dis. Mon 2008, 54, 763–768. [Google Scholar]
- Enright, M.C. The evolution of a resistant pathogen—The case of MRSA. Curr. Opin. Pharmacol 2003, 3, 474–479. [Google Scholar]
- Naimi, T.S.; LeDell, K.H.; Como-Sabetti, K.; Borchardt, S.M.; Boxrud, D.J.; Etienne, J.; Johnson, S.K.; Vandenesch, F.; Fridkin, S.; O’Boyle, C.; et al. Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. J. Am. Med. Assoc 2003, 290, 2976–2984. [Google Scholar]
- Klein, E.; Smith, D.L.; Laxminarayan, R. Community-associated methicillin-resistant Staphylococcus aureus in outpatients, United States, 1999–2006. Emerg. Infect. Dis 2009, 15, 1925–1930. [Google Scholar]
- Ghuysen, J.M. Molecular structures of penicillin-binding proteins and β-lactamases. Trends. Microbiol 1994, 2, 372–380. [Google Scholar]
- Katayama, Y.; Ito, T.; Hiramatsu, K. A new class of genetic element, staphylococcus cassette chromosome mec, encodes methicillin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother 2000, 44, 1549–1555. [Google Scholar]
- Hiramatsu, K.; Cui, L.; Kuroda, M.; Ito, T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends. Microbiol 2001, 9, 486–493. [Google Scholar]
- Diekema, D.J.; Pfaller, M.A.; Schmitz, F.J.; Smayevsky, J.; Bell, J.; Jones, R.N.; Beach, M.; Group, S.P. Survey of infections due to Staphylococcus species: Frequency of occurrence and antimicrobial susceptibility of isolates collected in the United States, Canada, Latin America, Europe, and the Western Pacific region for the SENTRY Antimicrobial Surveillance Program, 1997–1999. Clin. Infect. Dis 2001, 32, S114–S132. [Google Scholar]
- Centers for Disease Control and Prevention. Staphylococcus aureus resistant to vancomycin—United States, 2002. MMWR Morb. Mortal. Wkly. Rep 2002, 51, 565–567.
- Hiramatsu, K.; Hanaki, H.; Ino, T.; Yabuta, K.; Oguri, T.; Tenover, F.C. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother 1997, 40, 135–136. [Google Scholar]
- Cho, J.S.; Xuan, C.; Miller, L.S. Lucky number seven: RNase 7 can prevent Staphylococcus aureus skin colonization. J. Invest. Dermatol 2010, 130, 2703–2706. [Google Scholar]
- Otto, M. Staphylococcus colonization of the skin and antimicrobial peptides. Expert. Rev. Dermatol 2010, 5, 183–195. [Google Scholar]
- Schauber, J.; Gallo, R.L. Antimicrobial peptides and the skin immune defense system. J. Allergy. Clin. Immunol 2009, 124, R13–R18. [Google Scholar]
- Sahl, H.G.; Pag, U.; Bonness, S.; Wagner, S.; Antcheva, N.; Tossi, A. Mammalian defensins: Structures and mechanism of antibiotic activity. J. Leukoc. Biol 2005, 77, 466–475. [Google Scholar]
- Miller, L.S.; Sorensen, O.E.; Liu, P.T.; Jalian, H.R.; Eshtiaghpour, D.; Behmanesh, B.E.; Chung, W.; Starner, T.D.; Kim, J.; Sieling, P.A.; et al. TGF-α regulates TLR expression and function on epidermal keratinocytes. J. Immunol 2005, 174, 6137–6143. [Google Scholar]
- Sorensen, O.E.; Thapa, D.R.; Roupe, K.M.; Valore, E.V.; Sjobring, U.; Roberts, A.A.; Schmidtchen, A.; Ganz, T. Injury-induced innate immune response in human skin mediated by transactivation of the epidermal growth factor receptor. J. Clin. Invest 2006, 116, 1878–1885. [Google Scholar]
- Lehrer, R.I. Multispecific myeloid defensins. Curr. Opin. Hematol 2007, 14, 16–21. [Google Scholar]
- Ericksen, B.; Wu, Z.; Lu, W.; Lehrer, R.I. Antibacterial activity and specificity of the six human α-defensins. Antimicrob. Agents Chemother 2005, 49, 269–275. [Google Scholar]
- Peschel, A.; Jack, R.W.; Otto, M.; Collins, L.V.; Staubitz, P.; Nicholson, G.; Kalbacher, H.; Nieuwenhuizen, W.F.; Jung, G.; Tarkowski, A.; et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med 2001, 193, 1067–1076. [Google Scholar]
- Jin, T.; Bokarewa, M.; Foster, T.; Mitchell, J.; Higgins, J.; Tarkowski, A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J. Immunol 2004, 172, 1169–1176. [Google Scholar]
- Ernst, C.M.; Staubitz, P.; Mishra, N.N.; Yang, S.J.; Hornig, G.; Kalbacher, H.; Bayer, A.S.; Kraus, D.; Peschel, A. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog 2009, 5, e1000660. [Google Scholar]
- Jann, N.J.; Schmaler, M.; Kristian, S.A.; Radek, K.A.; Gallo, R.L.; Nizet, V.; Peschel, A.; Landmann, R. Neutrophil antimicrobial defense against Staphylococcus aureus is mediated by phagolysosomal but not extracellular trap-associated cathelicidin. J. Leukoc. Biol 2009, 86, 1159–1169. [Google Scholar]
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Gotz, F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem 1999, 274, 8405–8410. [Google Scholar]
- Dinulos, J.G.; Mentele, L.; Fredericks, L.P.; Dale, B.A.; Darmstadt, G.L. Keratinocyte expression of human β-defensin 2 following bacterial infection: Role in cutaneous host defense. Clin. Diagn. Lab. Immunol 2003, 10, 161–166. [Google Scholar]
- Sayama, K.; Komatsuzawa, H.; Yamasaki, K.; Shirakata, Y.; Hanakawa, Y.; Ouhara, K.; Tokumaru, S.; Dai, X.; Tohyama, M.; Ten Dijke, P.; et al. New mechanisms of skin innate immunity: ASK1-mediated keratinocyte differentiation regulates the expression of β-defensins, LL37, and TLR2. Eur. J. Immunol 2005, 35, 1886–1895. [Google Scholar]
- Simanski, M.; Glaser, R.; Koten, B.; Meyer-Hoffert, U.; Wanner, S.; Weidenmaier, C.; Peschel, A.; Harder, J. Staphylococcus aureus subverts cutaneous defense by d-alanylation of teichoic acids. Exp. Dermatol 2013, 22, 294–296. [Google Scholar]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. Isolation and characterization of human β-defensin 3, a novel human inducible peptide antibiotic. J. Biol. Chem 2001, 276, 5707–5713. [Google Scholar]
- Menzies, B.E.; Kenoyer, A. Signal transduction and nuclear responses in Staphylococcus aureus-induced expression of human β-defensin 3 in skin keratinocytes. Infect. Immun 2006, 74, 6847–6854. [Google Scholar]
- Sumikawa, Y.; Asada, H.; Hoshino, K.; Azukizawa, H.; Katayama, I.; Akira, S.; Itami, S. Induction of β-defensin 3 in keratinocytes stimulated by bacterial lipopeptides through Toll-like receptor 2. Microbes. Infect 2006, 8, 1513–1521. [Google Scholar]
- Zanger, P.; Holzer, J.; Schleucher, R.; Scherbaum, H.; Schittek, B.; Gabrysch, S. Severity of Staphylococcus aureus infection of the skin is associated with inducibility of human β-defensin 3 but not human β-defensin 2. Infect. Immun 2010, 78, 3112–3117. [Google Scholar]
- Garcia, J.R.; Krause, A.; Schulz, S.; Rodriguez-Jimenez, F.J.; Kluver, E.; Adermann, K.; Forssmann, U.; Frimpong-Boateng, A.; Bals, R.; Forssmann, W.G. Human β-defensin 4: A novel inducible peptide with a specific salt-sensitive spectrum of antimicrobial activity. FASEB J 2001, 15, 1819–1821. [Google Scholar]
- Rieg, S.; Steffen, H.; Seeber, S.; Humeny, A.; Kalbacher, H.; Dietz, K.; Garbe, C.; Schittek, B. Deficiency of dermcidin-derived antimicrobial peptides in sweat of patients with atopic dermatitis correlates with an impaired innate defense of human skin in vivo. J. Immunol. 2005, 174, 8003–8010. [Google Scholar]
- Steffen, H.; Rieg, S.; Wiedemann, I.; Kalbacher, H.; Deeg, M.; Sahl, H.G.; Peschel, A.; Gotz, F.; Garbe, C.; Schittek, B. Naturally processed dermcidin-derived peptides do not permeabilize bacterial membranes and kill microorganisms irrespective of their charge. Antimicrob. Agents Chemother 2006, 50, 2608–2620. [Google Scholar]
- Lai, Y.; Villaruz, A.E.; Li, M.; Cha, D.J.; Sturdevant, D.E.; Otto, M. The human anionic antimicrobial peptide dermcidin induces proteolytic defence mechanisms in staphylococci. Mol. Microbiol 2007, 63, 497–506. [Google Scholar]
- Grigat, J.; Soruri, A.; Forssmann, U.; Riggert, J.; Zwirner, J. Chemoattraction of macrophages, T lymphocytes, and mast cells is evolutionarily conserved within the human α-defensin family. J. Immunol 2007, 179, 3958–3965. [Google Scholar]
- Rohrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Human β-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J. Immunol 2010, 184, 6688–6694. [Google Scholar]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schroder, J.M.; Wang, J.M.; Howard, O.M.; et al. β-Defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 1999, 286, 525–528. [Google Scholar]
- De, Y.; Chen, Q.; Schmidt, A.P.; Anderson, G.M.; Wang, J.M.; Wooters, J.; Oppenheim, J.J.; Chertov, O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med 2000, 192, 1069–1074. [Google Scholar]
- Tjabringa, G.S.; Ninaber, D.K.; Drijfhout, J.W.; Rabe, K.F.; Hiemstra, P.S. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. Int. Arch. Allergy. Immunol 2006, 140, 103–112. [Google Scholar]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol 2003, 3, 710–720. [Google Scholar]
- Harder, J.; Meyer-Hoffert, U.; Wehkamp, K.; Schwichtenberg, L.; Schroder, J.M. Differential gene induction of human β-defensins (hBD-1, -2, -3, and -4) in keratinocytes is inhibited by retinoic acid. J. Invest. Dermatol 2004, 123, 522–529. [Google Scholar]
- Miller, L.S.; Modlin, R.L. Human keratinocyte Toll-like receptors promote distinct immune responses. J. Invest. Dermatol 2007, 127, 262–263. [Google Scholar]
- Yang, D.; Chertov, O.; Oppenheim, J.J. Participation of mammalian defensins and cathelicidins in anti-microbial immunity: Receptors and activities of human defensins and cathelicidin (LL-37). J. Leukoc. Biol 2001, 69, 691–697. [Google Scholar]
- Zanetti, M. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol 2004, 75, 39–48. [Google Scholar]
- Murakami, M.; Ohtake, T.; Dorschner, R.A.; Schittek, B.; Garbe, C.; Gallo, R.L. Cathelicidin anti-microbial peptide expression in sweat, an innate defense system for the skin. J. Invest. Dermatol 2002, 119, 1090–1095. [Google Scholar]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar]
- Schauber, J.; Dorschner, R.A.; Coda, A.B.; Buchau, A.S.; Liu, P.T.; Kiken, D.; Helfrich, Y.R.; Kang, S.; Elalieh, H.Z.; Steinmeyer, A.; et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J. Clin. Invest 2007, 117, 803–811. [Google Scholar]
- Wang, T.T.; Nestel, F.P.; Bourdeau, V.; Nagai, Y.; Wang, Q.; Liao, J.; Tavera-Mendoza, L.; Lin, R.; Hanrahan, J.W.; Mader, S.; et al. Cutting edge: 1,25-Dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol 2004, 173, 2909–2912. [Google Scholar]
- Zhang, J.; Dyer, K.D.; Rosenberg, H.F. Human RNase 7: A new cationic ribonuclease of the RNase A superfamily. Nucleic. Acids. Res 2003, 31, 602–607. [Google Scholar]
- Rieg, S.; Garbe, C.; Sauer, B.; Kalbacher, H.; Schittek, B. Dermcidin is constitutively produced by eccrine sweat glands and is not induced in epidermal cells under inflammatory skin conditions. Br. J. Dermatol 2004, 151, 534–539. [Google Scholar]
- Schittek, B.; Hipfel, R.; Sauer, B.; Bauer, J.; Kalbacher, H.; Stevanovic, S.; Schirle, M.; Schroeder, K.; Blin, N.; Meier, F.; et al. Dermcidin: A novel human antibiotic peptide secreted by sweat glands. Nat. Immunol 2001, 2, 1133–1137. [Google Scholar]
- Hruz, P.; Zinkernagel, A.S.; Jenikova, G.; Botwin, G.J.; Hugot, J.P.; Karin, M.; Nizet, V.; Eckmann, L. NOD2 contributes to cutaneous defense against Staphylococcus aureus through α-toxin-dependent innate immune activation. Proc. Natl. Acad. Sci. USA 2009, 106, 12873–12878. [Google Scholar]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar]
- Kim, M.H.; Granick, J.L.; Kwok, C.; Walker, N.J.; Borjesson, D.L.; Curry, F.R.; Miller, L.S.; Simon, S.I. Neutrophil survival and c-kit+-progenitor proliferation in Staphylococcus aureus-infected skin wounds promote resolution. Blood 2011, 117, 3343–3352. [Google Scholar]
- Molne, L.; Verdrengh, M.; Tarkowski, A. Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect. Immun 2000, 68, 6162–6167. [Google Scholar]
- Miller, L.S.; Cho, J.S. Immunity against Staphylococcus aureus cutaneous infections. Nat. Rev. Immunol 2011, 11, 505–518. [Google Scholar]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol 2010, 10, 479–489. [Google Scholar]
- Kraus, D.; Peschel, A. Staphylococcus aureus evasion of innate antimicrobial defense. Future Microbiol 2008, 3, 437–451. [Google Scholar]
- Huijbregts, R.P.; de Kroon, A.I.; de Kruijff, B. Topology and transport of membrane lipids in bacteria. Biochim. Biophys. Acta 2000, 1469, 43–61. [Google Scholar]
- Koprivnjak, T.; Peschel, A.; Gelb, M.H.; Liang, N.S.; Weiss, J.P. Role of charge properties of bacterial envelope in bactericidal action of human group IIA phospholipase A2 against Staphylococcus aureus. J. Biol. Chem 2002, 277, 47636–47644. [Google Scholar]
- Kristian, S.A.; Durr, M.; van Strijp, J.A.; Neumeister, B.; Peschel, A. MprF-mediated lysinylation of phospholipids in Staphylococcus aureus leads to protection against oxygen-independent neutrophil killing. Infect. Immun 2003, 71, 546–549. [Google Scholar]
- Staubitz, P.; Neumann, H.; Schneider, T.; Wiedemann, I.; Peschel, A. MprF-mediated biosynthesis of lysylphosphatidylglycerol, an important determinant in staphylococcal defensin resistance. FEMS Microbiol. Lett 2004, 231, 67–71. [Google Scholar]
- Neuhaus, F.C.; Baddiley, J. A continuum of anionic charge: Structures and functions of d-alanyl-teichoic acids in gram-positive bacteria. Microbiol. Mol. Biol. Rev 2003, 67, 686–723. [Google Scholar]
- Oku, Y.; Kurokawa, K.; Ichihashi, N.; Sekimizu, K. Characterization of the Staphylococcus aureus mprF gene, involved in lysinylation of phosphatidylglycerol. Microbiology 2004, 150, 45–51. [Google Scholar]
- Collins, L.V.; Kristian, S.A.; Weidenmaier, C.; Faigle, M.; van Kessel, K.P.; van Strijp, J.A.; Gotz, F.; Neumeister, B.; Peschel, A. Staphylococcus aureus strains lacking d-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J. Infect. Dis 2002, 186, 214–219. [Google Scholar]
- Kristian, S.A.; Lauth, X.; Nizet, V.; Goetz, F.; Neumeister, B.; Peschel, A.; Landmann, R. Alanylation of teichoic acids protects Staphylococcus aureus against Toll-like receptor 2-dependent host defense in a mouse tissue cage infection model. J. Infect. Dis 2003, 188, 414–423. [Google Scholar]
- Weidenmaier, C.; Peschel, A.; Kempf, V.A.; Lucindo, N.; Yeaman, M.R.; Bayer, A.S. DltABCDand MprF-mediated cell envelope modifications of Staphylococcus aureus confer resistance to platelet microbicidal proteins and contribute to virulence in a rabbit endocarditis model. Infect. Immun 2005, 73, 8033–8038. [Google Scholar]
- Peschel, A.; Sahl, H.G. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol 2006, 4, 529–536. [Google Scholar]
- Ouhara, K.; Komatsuzawa, H.; Kawai, T.; Nishi, H.; Fujiwara, T.; Fujiue, Y.; Kuwabara, M.; Sayama, K.; Hashimoto, K.; Sugai, M. Increased resistance to cationic antimicrobial peptide LL-37 in methicillin-resistant strains of Staphylococcus aureus. J. Antimicrob. Chemother 2008, 61, 1266–1269. [Google Scholar]
- Friedman, L.; Alder, J.D.; Silverman, J.A. Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother 2006, 50, 2137–2145. [Google Scholar]
- Jones, T.; Yeaman, M.R.; Sakoulas, G.; Yang, S.J.; Proctor, R.A.; Sahl, H.G.; Schrenzel, J.; Xiong, Y.Q.; Bayer, A.S. Failures in clinical treatment of Staphylococcus aureus Infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob. Agents Chemother 2008, 52, 269–278. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Peptides | Cellular source in the skin | Mechanism of S. aureus evasion | References |
|---|---|---|---|
| α-Defensins | Neutrophils | Staphylokinase, MprF, dltABCD operon | [67–73] |
| hBD2 | Keratinocytes, macrophages, and dendritic cells | IsdA, dltABCD | [26,29,35,74–76] |
| hBD3 | Keratinocytes | dltABCD operon | [23,24,27,75–80] |
| hBD4 | Keratinocytes | Please check | [81] |
| LL-37 | Keratinocytes, macrophages, and neutrophils | IsdA, Aureolysin, MprF, dltABCD | [25,29,35,36,69,71–73,75] |
| Dermcidin | Sweat glands | Extracellular proteases, dltABCD operon | [82–84] |
| RNase 7 | Keratinocytes | dltABCD operon | [24,28,61,76] |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ryu, S.; Song, P.I.; Seo, C.H.; Cheong, H.; Park, Y. Colonization and Infection of the Skin by S. aureus: Immune System Evasion and the Response to Cationic Antimicrobial Peptides. Int. J. Mol. Sci. 2014, 15, 8753-8772. https://doi.org/10.3390/ijms15058753
Ryu S, Song PI, Seo CH, Cheong H, Park Y. Colonization and Infection of the Skin by S. aureus: Immune System Evasion and the Response to Cationic Antimicrobial Peptides. International Journal of Molecular Sciences. 2014; 15(5):8753-8772. https://doi.org/10.3390/ijms15058753
Chicago/Turabian StyleRyu, Sunhyo, Peter I. Song, Chang Ho Seo, Hyeonsook Cheong, and Yoonkyung Park. 2014. "Colonization and Infection of the Skin by S. aureus: Immune System Evasion and the Response to Cationic Antimicrobial Peptides" International Journal of Molecular Sciences 15, no. 5: 8753-8772. https://doi.org/10.3390/ijms15058753