Total Protein Extraction for Metaproteomics Analysis of Methane Producing Biofilm: The Effects of Detergents

Abstract
:1. Introduction
2. Results and Discussion
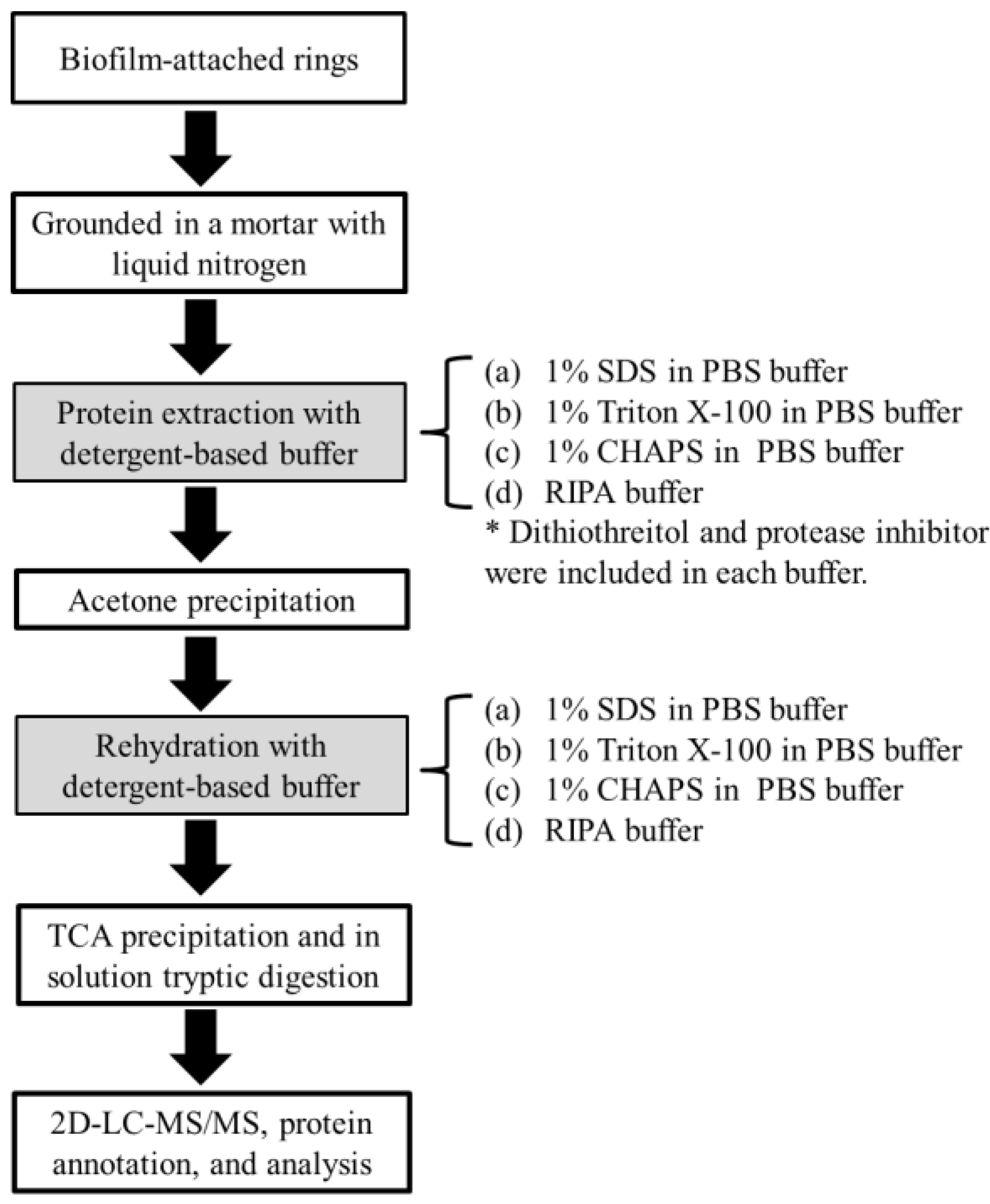
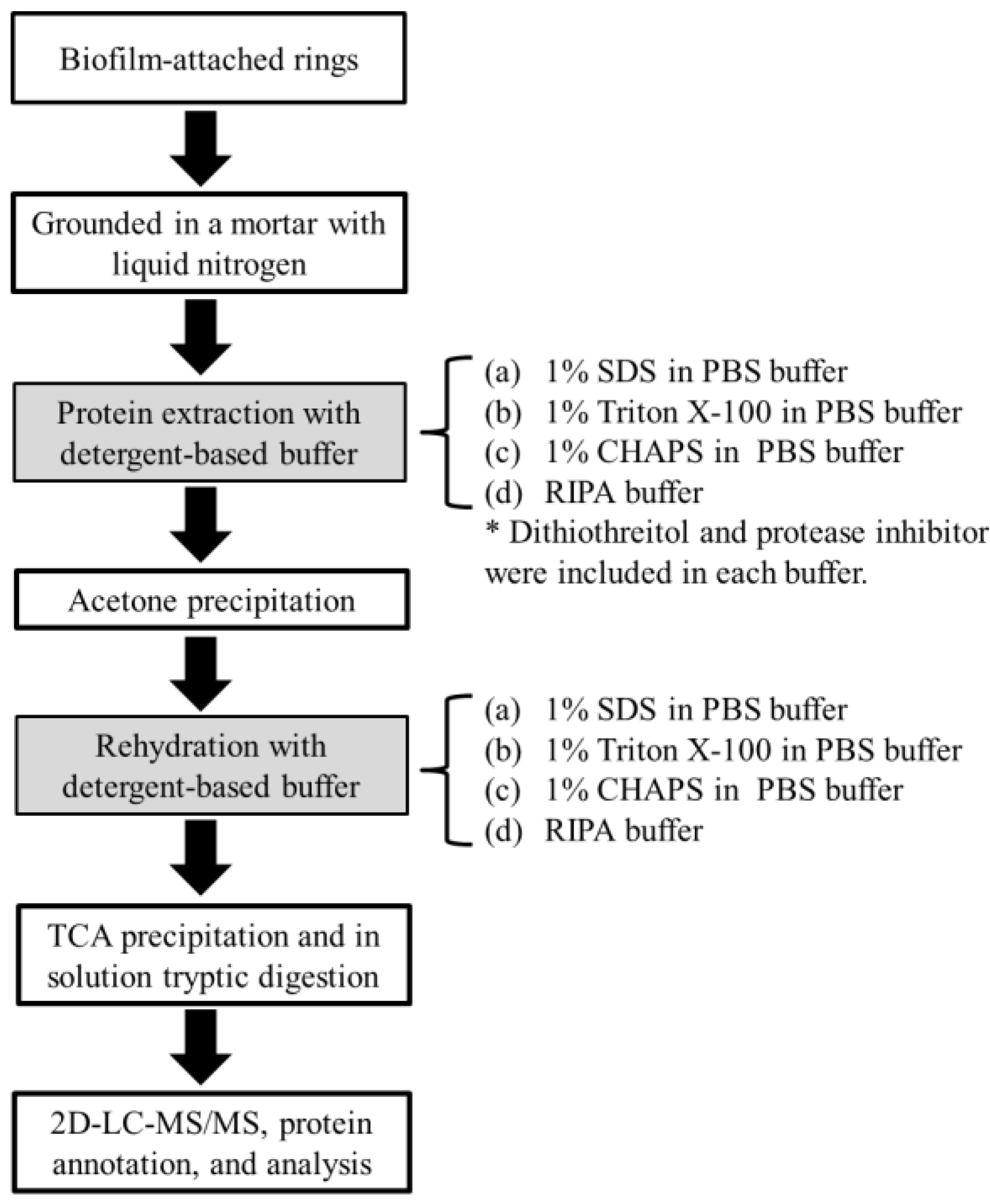
2.1. Yield and Number of Proteins Recovered from Methanogenic Biofilm

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analysis | Extraction Buffer Containing | |||
|---|---|---|---|---|
| SDS | Triton X-100 | CHAPS | RIPA | |
| Protein recovery (μg/g) | 1182.9 ± 196.6 | 884.8 ± 221.0 | 1065.4 ± 217.9 | 1056.1 ± 241.4 |
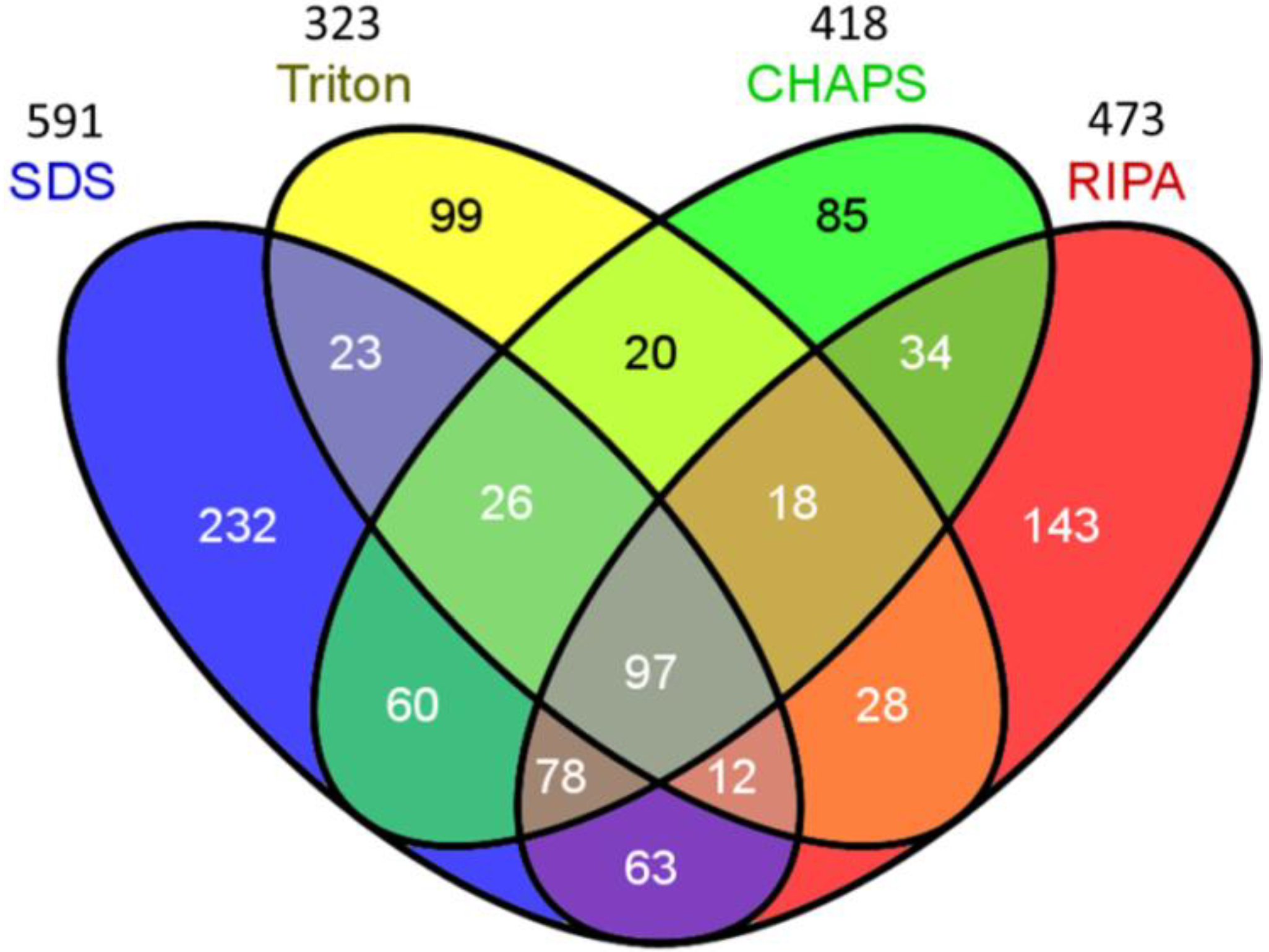
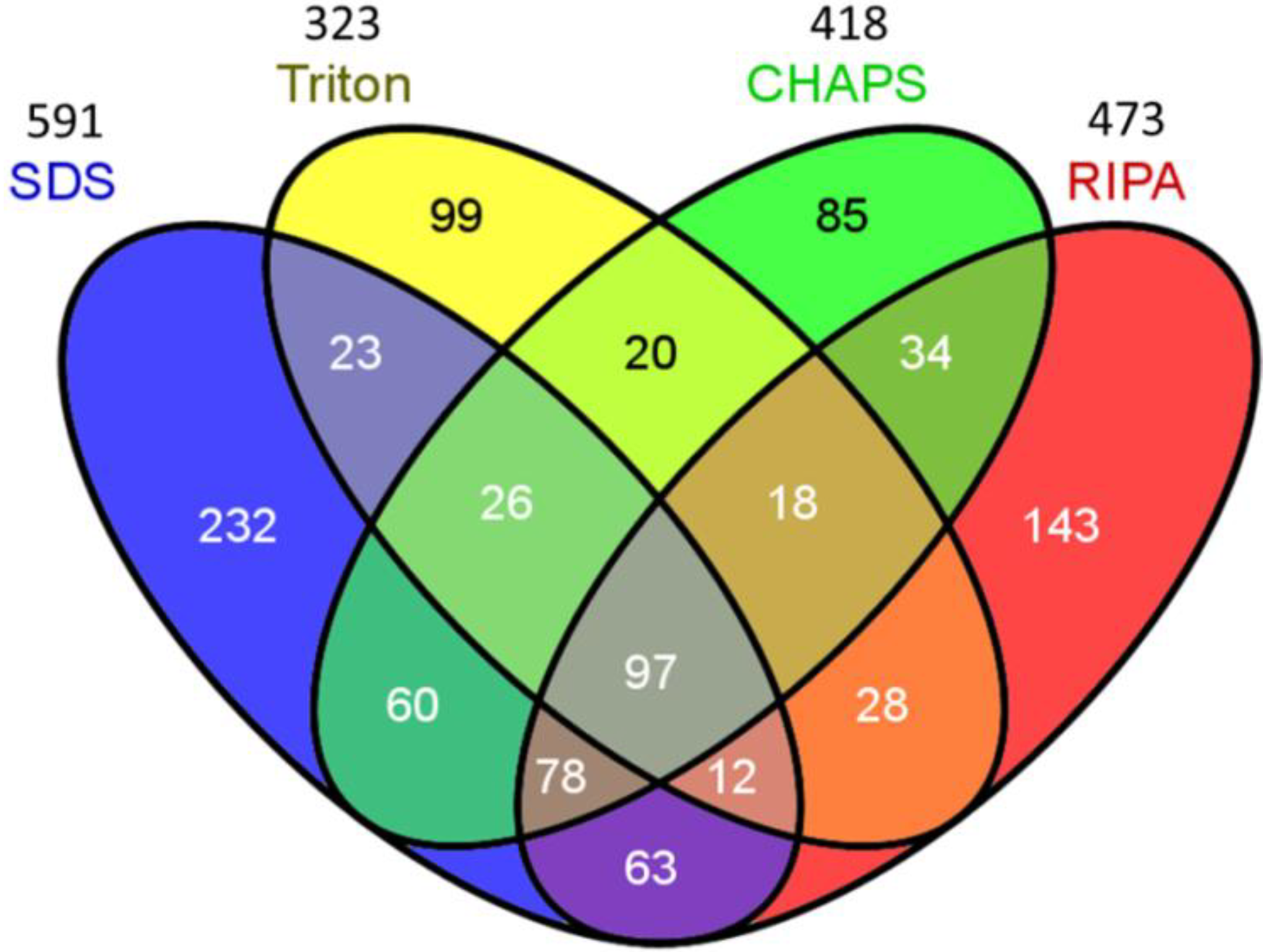
| Number of identified protein 1 | 591 | 323 | 418 | 473 |
| Proteins 2 | 1206 | 836 | 995 | 1121 |
| Unique peptide number | 1955 | 1061 | 1407 | 1395 |
| Unique spectra number | 2110 | 1130 | 1530 | 1545 |
| Proteins 3 | 477 | 317 | 439 | 447 |
| Unique peptide number | 1345 | 614 | 955 | 988 |
| Unique spectra number | 1476 | 663 | 1060 | 1109 |
| Membrane protein (% of total) 4 | 103 (17.5%) | 34 (10.6%) | 69 (16.5%) | 99 (21.0%) |
2.2. Venn Diagram Analysis
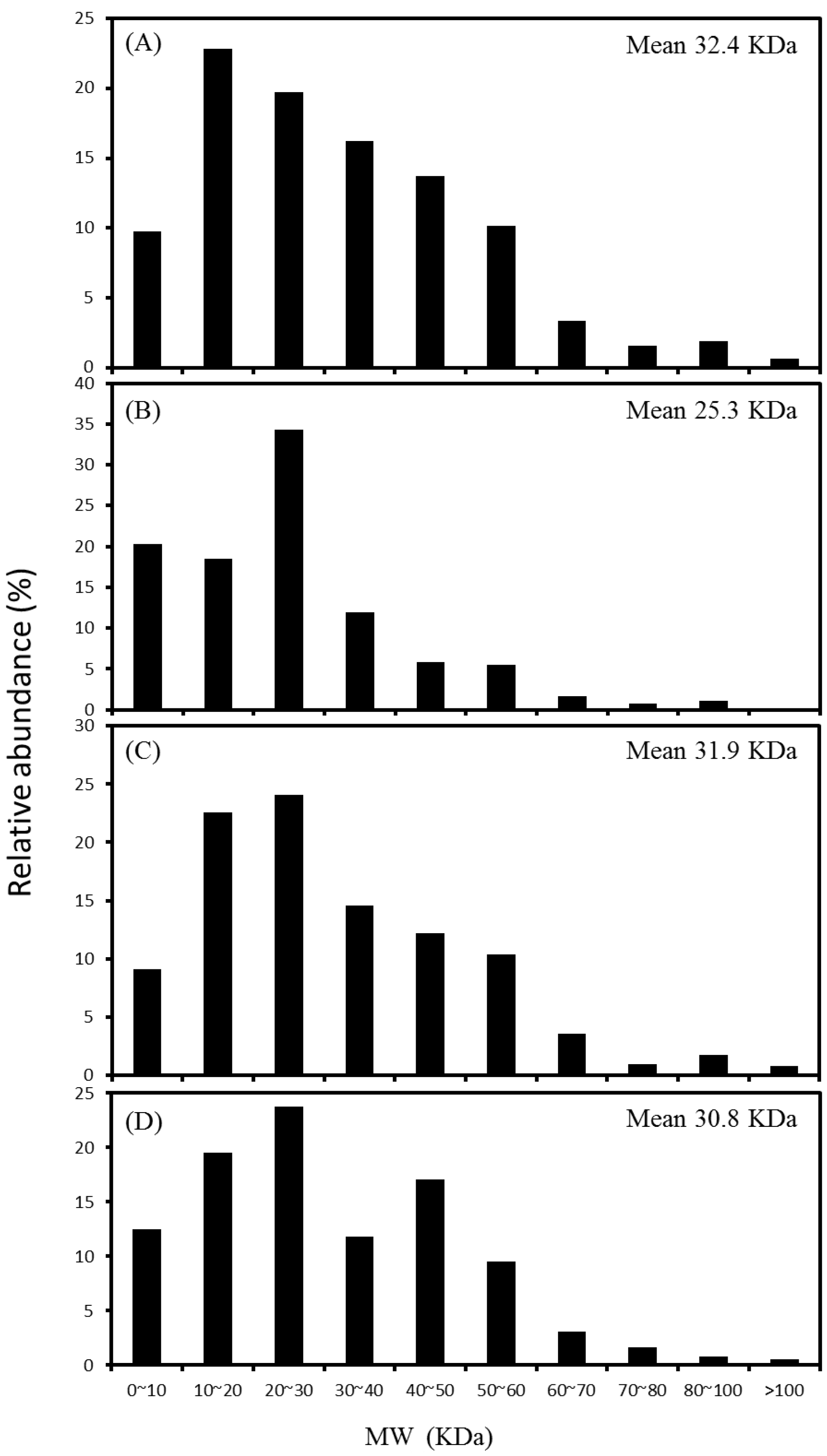
2.3. Distribution of Protein Molecular Weight

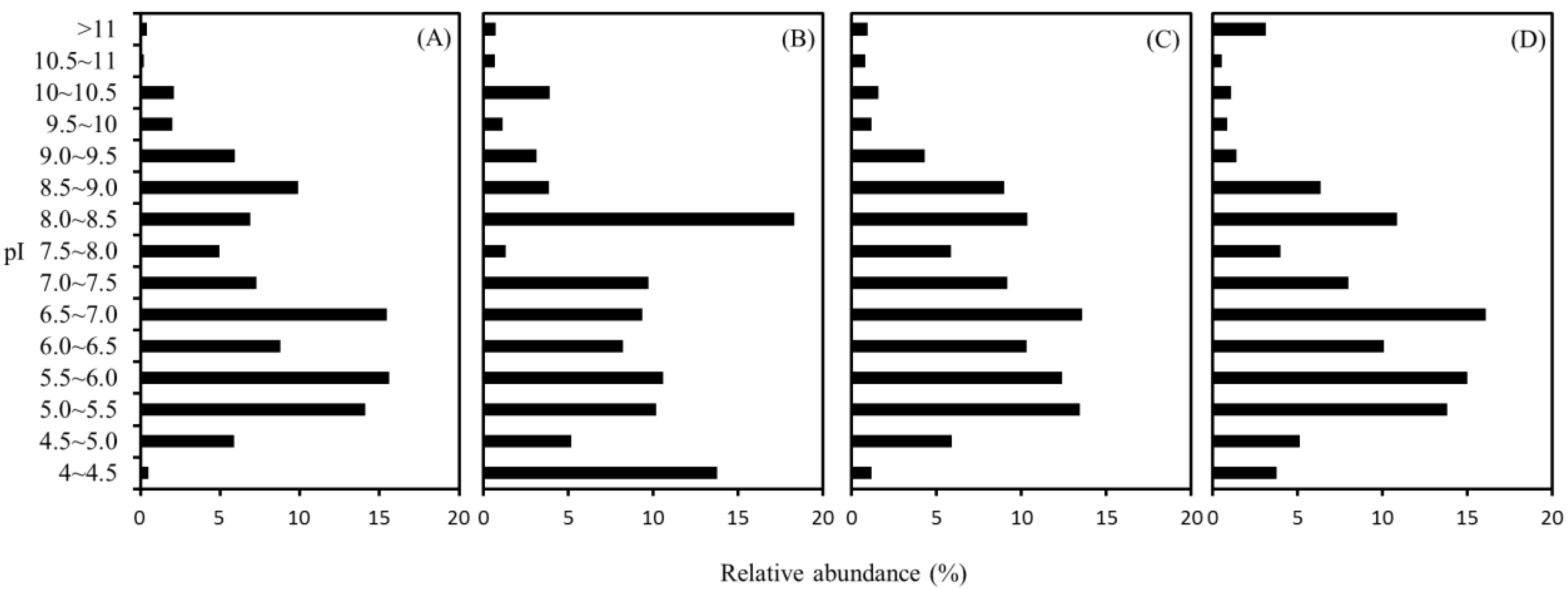
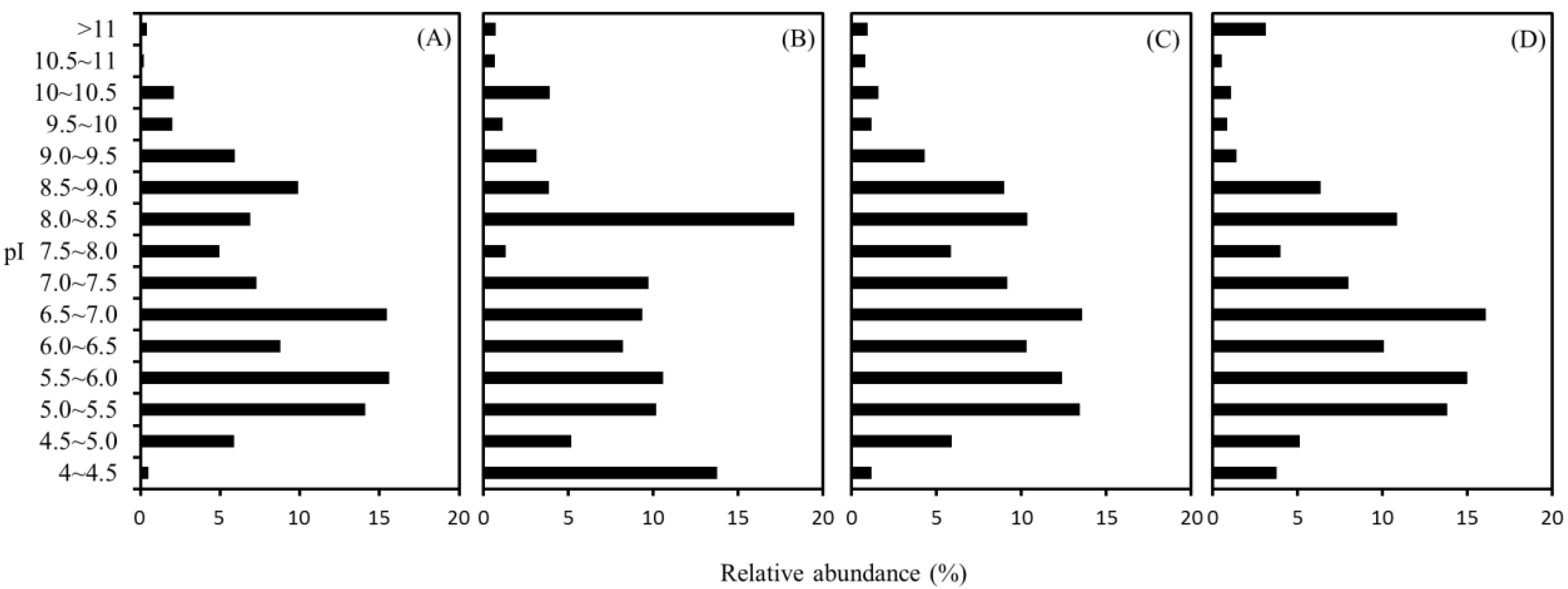
2.4. Distribution of Isoelectric Point (pI)
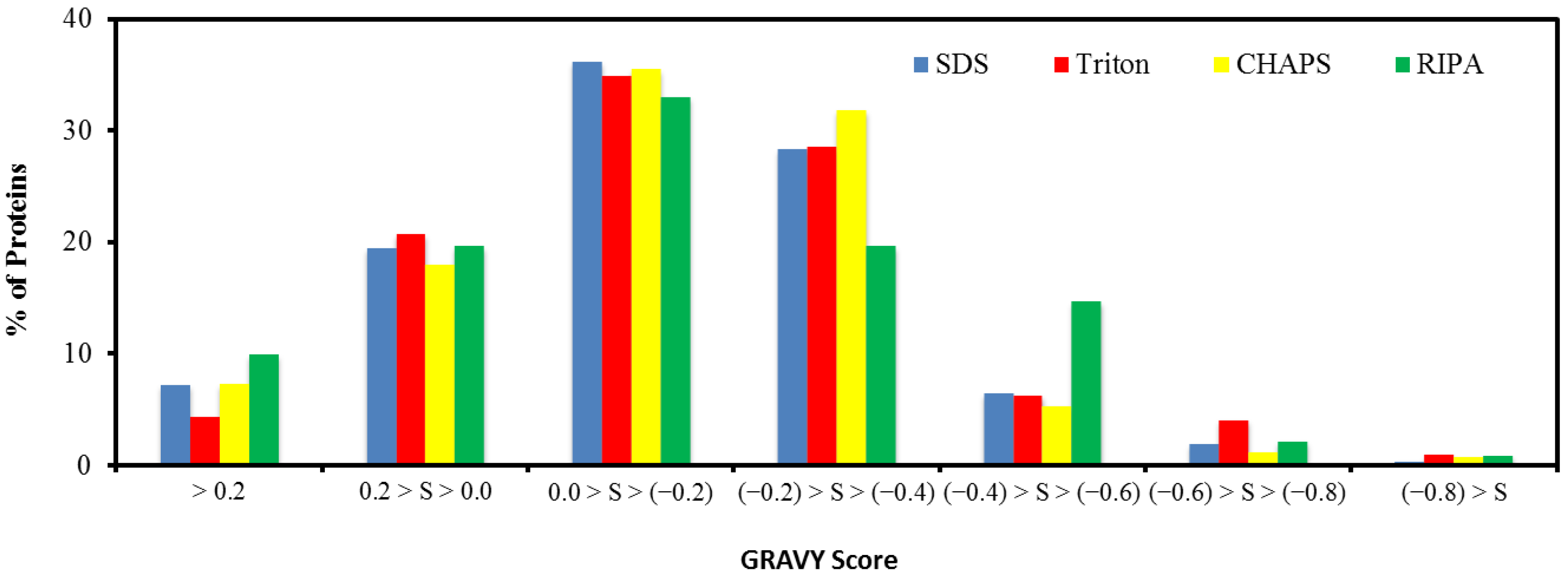
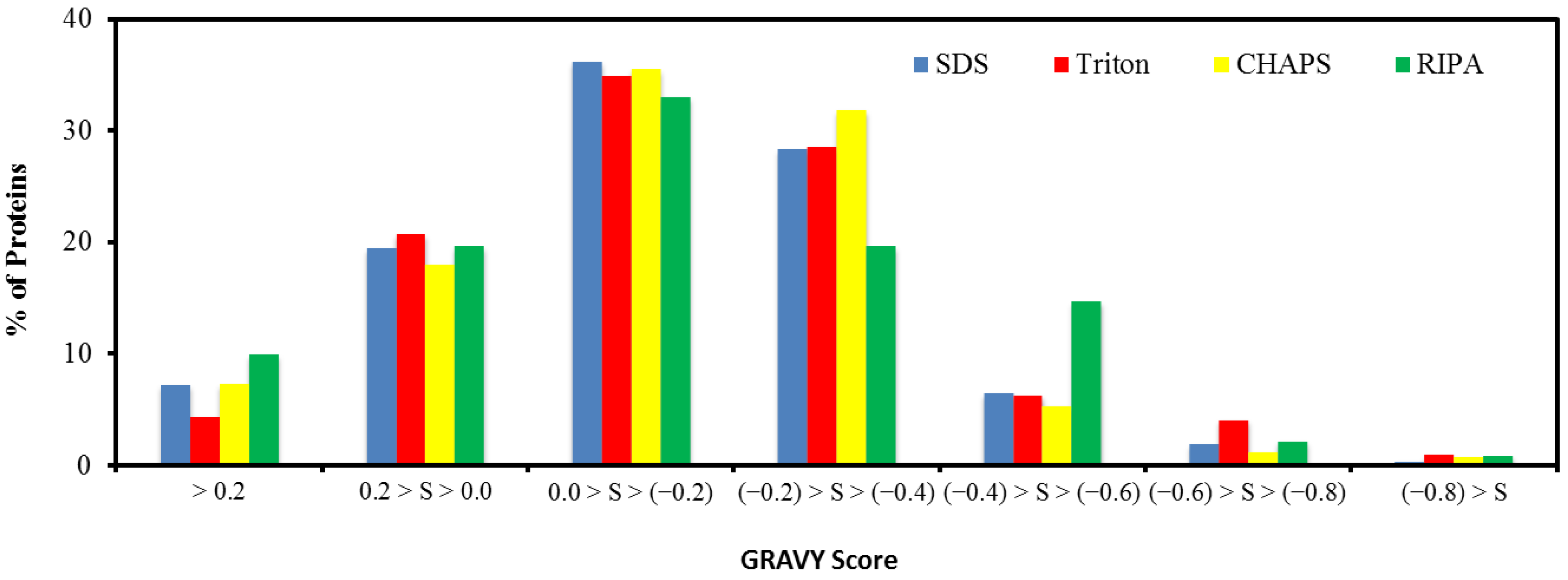
2.5. Hydrophobicity of Protein
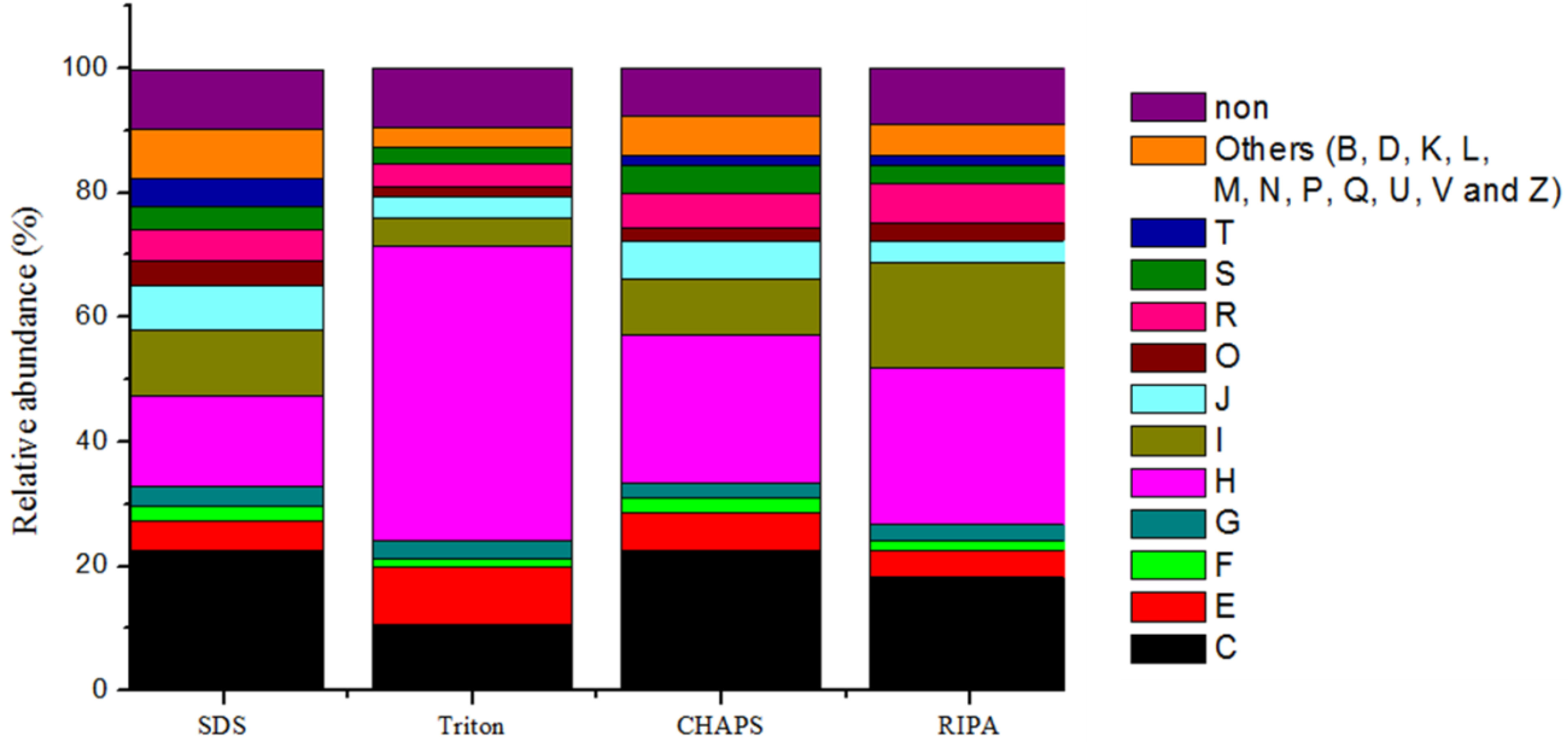
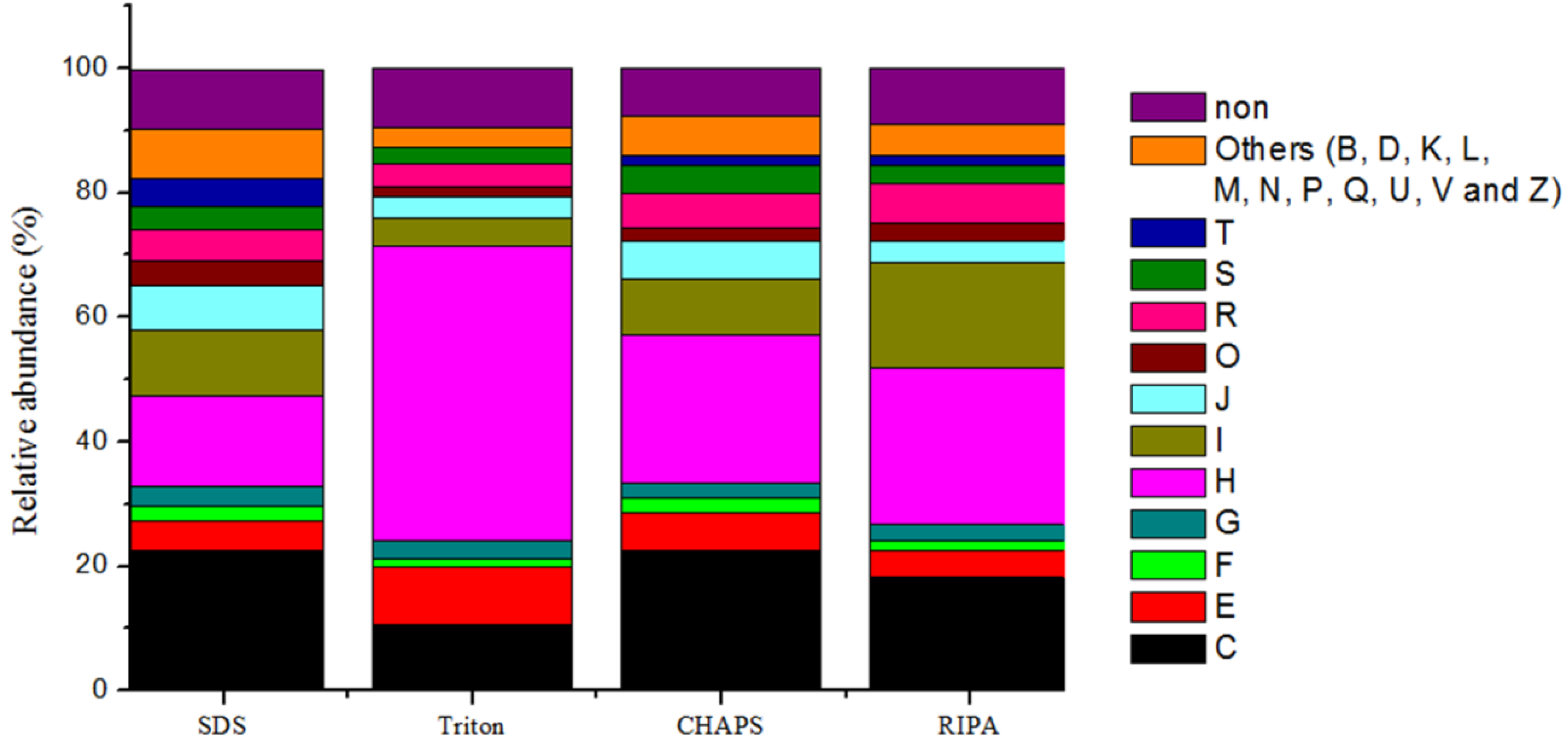
2.6. Expression of Microbial Functions
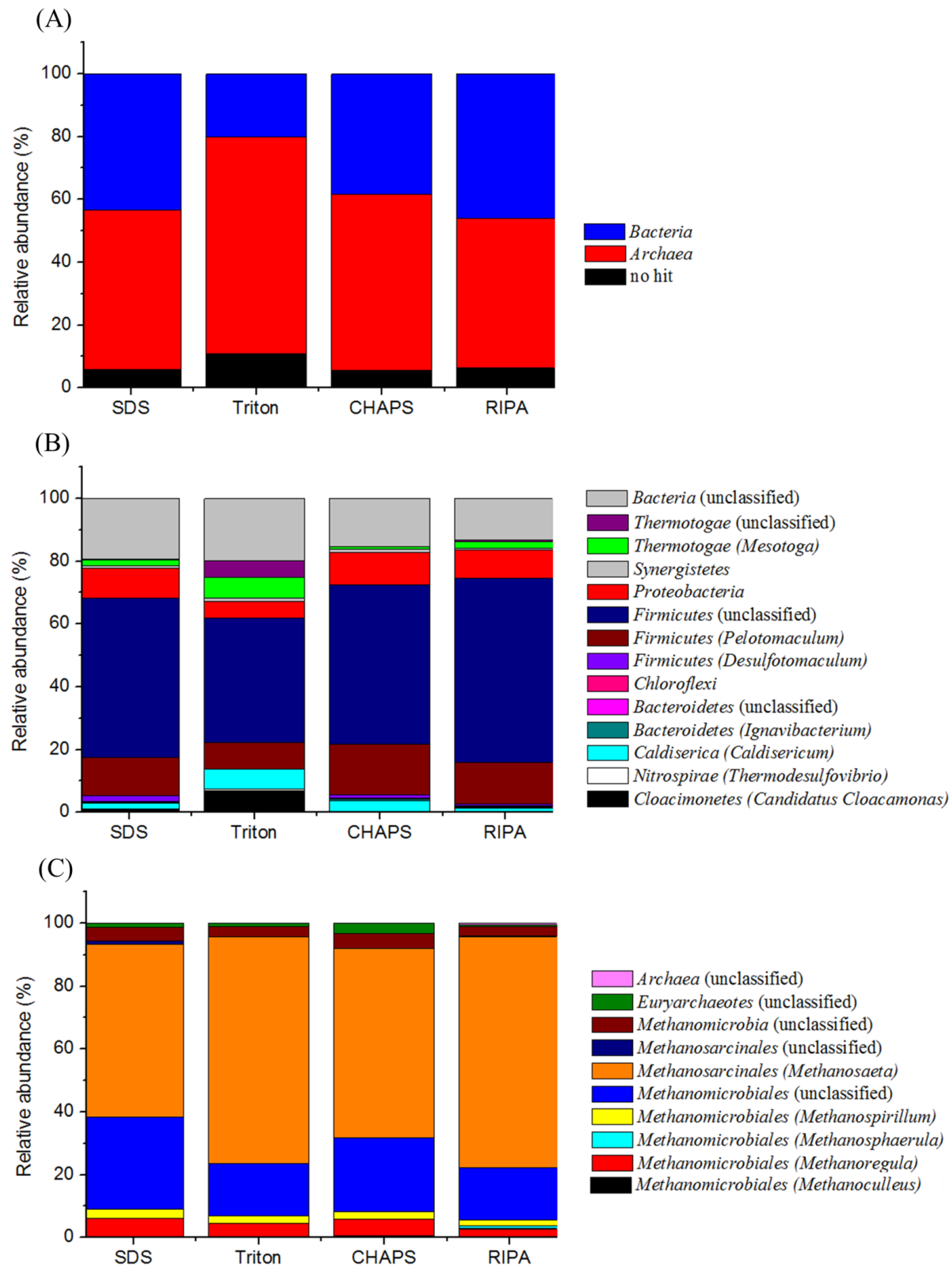
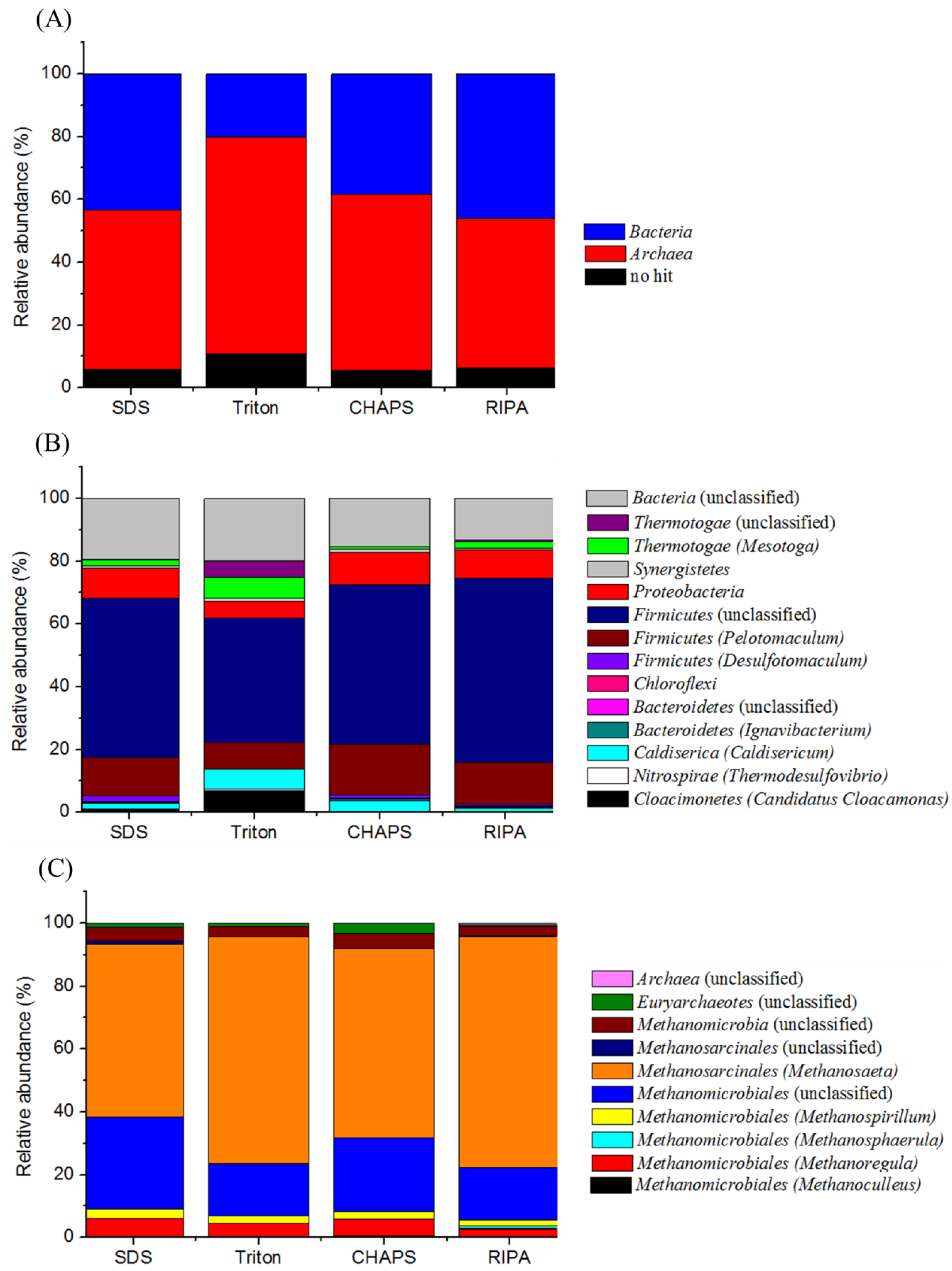
2.7. Proteomic-Based Microbial Community Structure
2.8. Clone Library Analysis of the 16S rRNA Gene Sequences
3. Experimental Section
3.1. Bioreactor and Sampling
3.2. Protein Extraction
3.3. In Solution Tryptic Digestion
3.4. On-Line Two Dimensional HPLC-MS/MS Analysis
3.5. Data Processing and Protein Identification
3.6. Protein Annotations and Analysis
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar]
- Kolmeder, C.A.; de Vos, W.M. Metaproteomics of our microbiome: Developing insight in function and activity in man and model systems. J. Proteomics 2014, 97, 3–16. [Google Scholar] [CrossRef]
- Stranger, B.E.; Stahl, E.A.; Raj, T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 2011, 187, 367–383. [Google Scholar] [CrossRef]
- Wu, J.H.; Wu, F.Y.; Chuang, H.P.; Chen, W.Y.; Huang, H.J.; Chen, S.H.; Liu, W.T. Community and proteomic analysis of methanogenic consortia degrading terephthalate. Appl. Environ. Microbiol. 2013, 79, 105–112. [Google Scholar] [CrossRef]
- Ram, R.J.; Verberkmoes, N.C.; Thelen, M.P.; Tyson, G.W.; Baker, B.J.; Blake, R.C., 2nd; Shah, M.; Hettich, R.L.; Banfield, J.F. Community proteomics of a natural microbial biofilm. Science 2005, 308, 1915–1920, doi:10.1126/science. 1109070. [Google Scholar]
- Keller, M.; Hettich, R. Environmental proteomics: a paradigm shift in characterizing microbial activities at the molecular level. Microbiol. Mol. Biol. Rev. 2009, 73, 62–70. [Google Scholar] [CrossRef]
- Leary, D.H.; Hervey, W.J., 4th; Deschamps, J.R.; Kusterbeck, A.W.; Vora, G.J. Which metaproteome? The impact of protein extraction bias on metaproteomic analyses. Mol. Cell. Probes 2013, 27, 193–199. [Google Scholar] [CrossRef]
- Shabbiri, K.; Botting, C.H.; Adnan, A.; Fuszard, M. Charting the cellular and extracellular proteome analysis of Brevibacterium linens DSM 20158 with unsequenced genome by mass spectrometry-driven sequence similarity searches. J. Proteomics 2013, 83, 99–118. [Google Scholar] [CrossRef]
- Phillips, N.J.; Steichen, C.T.; Schilling, B.; Post, D.M.; Niles, R.K.; Bair, T.B.; Falsetta, M.L.; Apicella, M.A.; Gibson, B.W. Proteomic analysis of Neisseria gonorrhoeae biofilms shows shift to anaerobic respiration and changes in nutrient transport and outermembrane proteins. PLoS One 2012, 7, e38303. [Google Scholar] [CrossRef]
- Giagnoni, L.; Magherini, F.; Landi, L.; Taghavi, S.; Modesti, A.; Bini, L.; Nannipieri, P.; van der Lelie, D.; Renella, G. Extraction of microbial proteome from soil: potential and limitations assessed through a model study. Eur. J. Soil Sci. 2011, 62, 74–81. [Google Scholar] [CrossRef]
- Wu, C.C.; MacCoss, M.J. Shotgun proteomics: Tools for the analysis of complex biological systems. Curr. Opin. Mol. Ther. 2002, 4, 242–250. [Google Scholar]
- Schneider, T.; Riedel, K. Environmental proteomics: analysis of structure and function of microbial communities. Proteomics 2010, 10, 785–798. [Google Scholar] [CrossRef]
- Abram, F.; Gunnigle, E.; O’Flaherty, V. Optimisation of protein extraction and 2-DE for metaproteomics of microbial communities from anaerobic wastewater treatment biofilms. Electrophoresis 2009, 30, 4149–4151. [Google Scholar] [CrossRef]
- Leary, D.H.; Hervey, W.J., 4th; Li, R.W.; Deschamps, J.R.; Kusterbeck, A.W.; Vora, G.J. Method development for metaproteomic analyses of marine biofilms. Anal. Chem 2012, 84, 4006–4013. [Google Scholar] [CrossRef]
- Arachea, B.T.; Sun, Z.; Potente, N.; Malik, R.; Isailovic, D.; Viola, R.E. Detergent selection for enhanced extraction of membrane proteins. Protein Expr. Purif. 2012, 86, 12–20. [Google Scholar] [CrossRef]
- Tanca, A.; Biosa, G.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Comparison of detergent-based sample preparation workflows for LTQ-Orbitrap analysis of the Escherichia coli proteome. Proteomics 2013, 13, 2597–2607. [Google Scholar] [CrossRef]
- Keiblinger, K.M.; Wilhartitz, I.C.; Schneider, T.; Roschitzki, B.; Schmid, E.; Eberl, L.; Riedel, K.; Zechmeister-Boltenstern, S. Soil metaproteomics: Comparative evaluation of protein extraction protocols. Soil Biol. Biochem. 2012, 54, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Lykidis, A.; Chen, C.L.; Tringe, S.G.; McHardy, A.C.; Copeland, A.; Kyrpides, N.C.; Hugenholtz, P.; Macarie, H.; Olmos, A.; Monroy, O.; et al. Multiple syntrophic interactions in a terephthalate-degrading methanogenic consortium. ISME J. 2011, 5, 122–130. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Wallin, E.; von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. [Google Scholar] [CrossRef]
- PSORTb. Available online: http://www.psort.org/ (accessed on 4 September 2013).
- Kiraga, J.; Mackiewicz, P.; Mackiewicz, D.; Kowalczuk, M.; Biecek, P.; Polak, N.; Smolarczyk, K.; Dudek, M.R.; Cebrat, S. The relationships between the isoelectric point and: length of proteins, taxonomy and ecology of organisms. BMC Genomics 2007, 8. [Google Scholar] [CrossRef]
- Schwartz, R.; Ting, C.S.; King, J. Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res. 2001, 11, 703–709. [Google Scholar]
- VanBogelen, R.A.; Schiller, E.E.; Thomas, J.D.; Neidhardt, F.C. Diagnosis of cellular states of microbial organisms using proteomics. Electrophoresis 1999, 20, 2149–2159. [Google Scholar] [CrossRef]
- Nelson, K.E.; Clayton, R.A.; Gill, S.R.; Gwinn, M.L.; Dodson, R.J.; Haft, D.H.; Hickey, E.K.; Peterson, J.D.; Nelson, W.C.; Ketchum, K.A.; et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 1999, 399, 323–329. [Google Scholar] [CrossRef]
- Bult, C.J.; White, O.; Olsen, G.J.; Zhou, L.; Fleischmann, R.D.; Sutton, G.G.; Blake, J.A.; FitzGerald, L.M.; Clayton, R.A.; Gocayne, J.D.; et al. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 1996, 273, 1058–1073. [Google Scholar]
- Lin, S.X.; Gangloff, A.; Huang, Y.W.; Xie, B.X. Electrophoresis of hydrophobic proteins. Anal. Chim. Acta 1999, 383, 101–107. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Huang, H.J.; Tsai, M.L.; Chen, Y.W.; Chen, S.H. Quantitative shot-gun proteomics and MS-based activity assay for revealing gender differences in enzyme contents for rat liver microsome. J. Proteomics 2011, 74, 2734–2744. [Google Scholar] [CrossRef]
- Wolters, D.A.; Washburn, M.P.; Yates, J.R., 3rd. An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 2001, 73, 5683–5690. [Google Scholar] [CrossRef]
- IMG/M. Available online: http://img.jgi.doe.gov (accessed on 3 December 2012).
- NCBI/CCD. Available online: http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml (accessed on 30 August 2013).
- Tatusov, R.L.; Natale, D.A.; Garkavtsev, I.V.; Tatusova, T.A.; Shankavaram, U.T.; Rao, B.S.; Kiryutin, B.; Galperin, M.Y.; Fedorova, N.D.; Koonin, E.V. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001, 29, 22–28. [Google Scholar] [CrossRef]
- Florens, L.; Carozza, M.J.; Swanson, S.K.; Fournier, M.; Coleman, M.K.; Workman, J.L.; Washburn, M.P. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 2006, 40, 303–311. [Google Scholar] [CrossRef]
- An Interactive Tool for Comparing Lists with Venn Diagrams. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 7 August 2013).
- Gravy Calculator. Available online: http://www.gravy-calculator.de (accessed on 29 July 2013).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, H.-J.; Chen, W.-Y.; Wu, J.-H. Total Protein Extraction for Metaproteomics Analysis of Methane Producing Biofilm: The Effects of Detergents. Int. J. Mol. Sci. 2014, 15, 10169-10184. https://doi.org/10.3390/ijms150610169
Huang H-J, Chen W-Y, Wu J-H. Total Protein Extraction for Metaproteomics Analysis of Methane Producing Biofilm: The Effects of Detergents. International Journal of Molecular Sciences. 2014; 15(6):10169-10184. https://doi.org/10.3390/ijms150610169
Chicago/Turabian StyleHuang, Hung-Jen, Wei-Yu Chen, and Jer-Horng Wu. 2014. "Total Protein Extraction for Metaproteomics Analysis of Methane Producing Biofilm: The Effects of Detergents" International Journal of Molecular Sciences 15, no. 6: 10169-10184. https://doi.org/10.3390/ijms150610169