Activation of mGluR5 Attenuates NMDA-Induced Neurotoxicity through Disruption of the NMDAR-PSD-95 Complex and Preservation of Mitochondrial Function in Differentiated PC12 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Disscussion
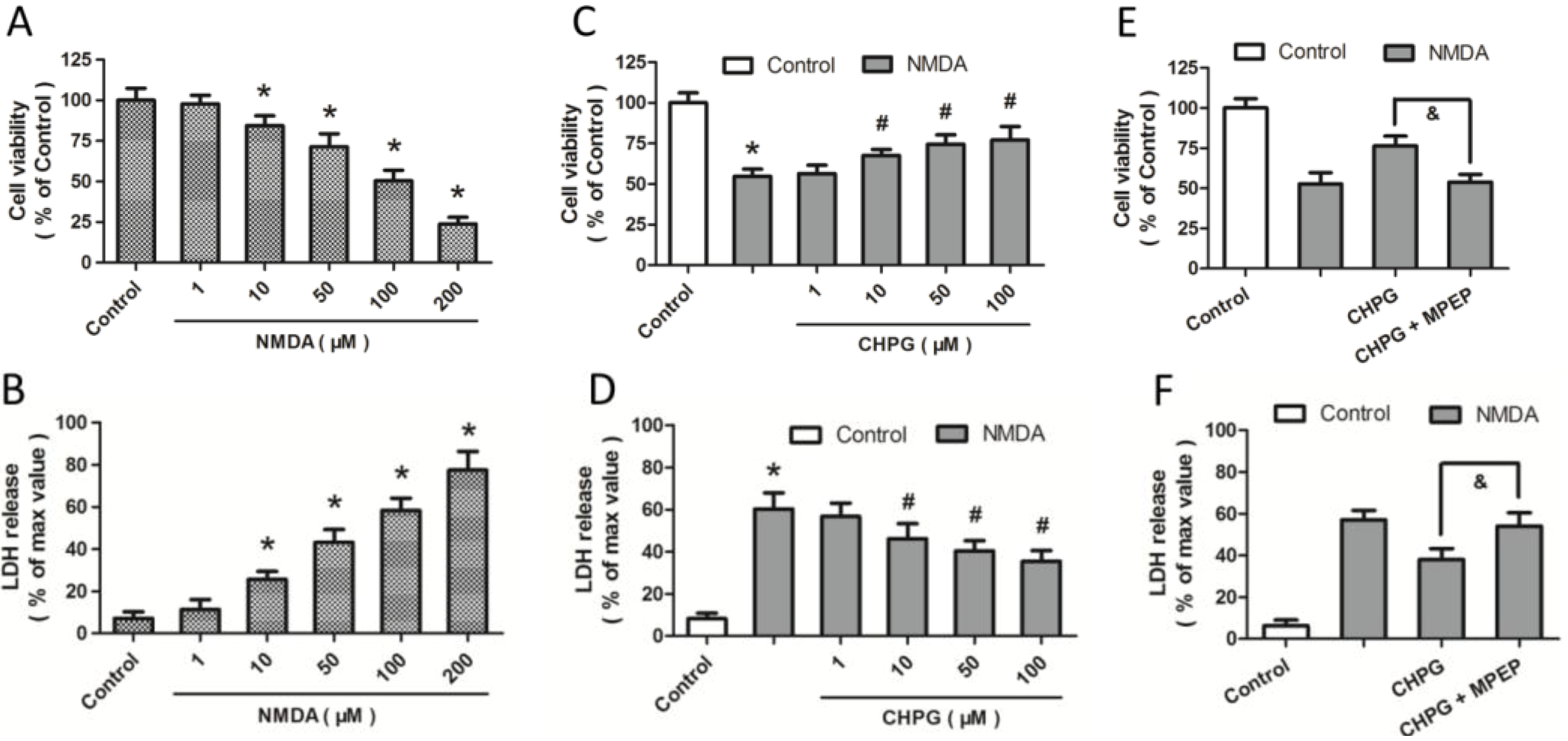
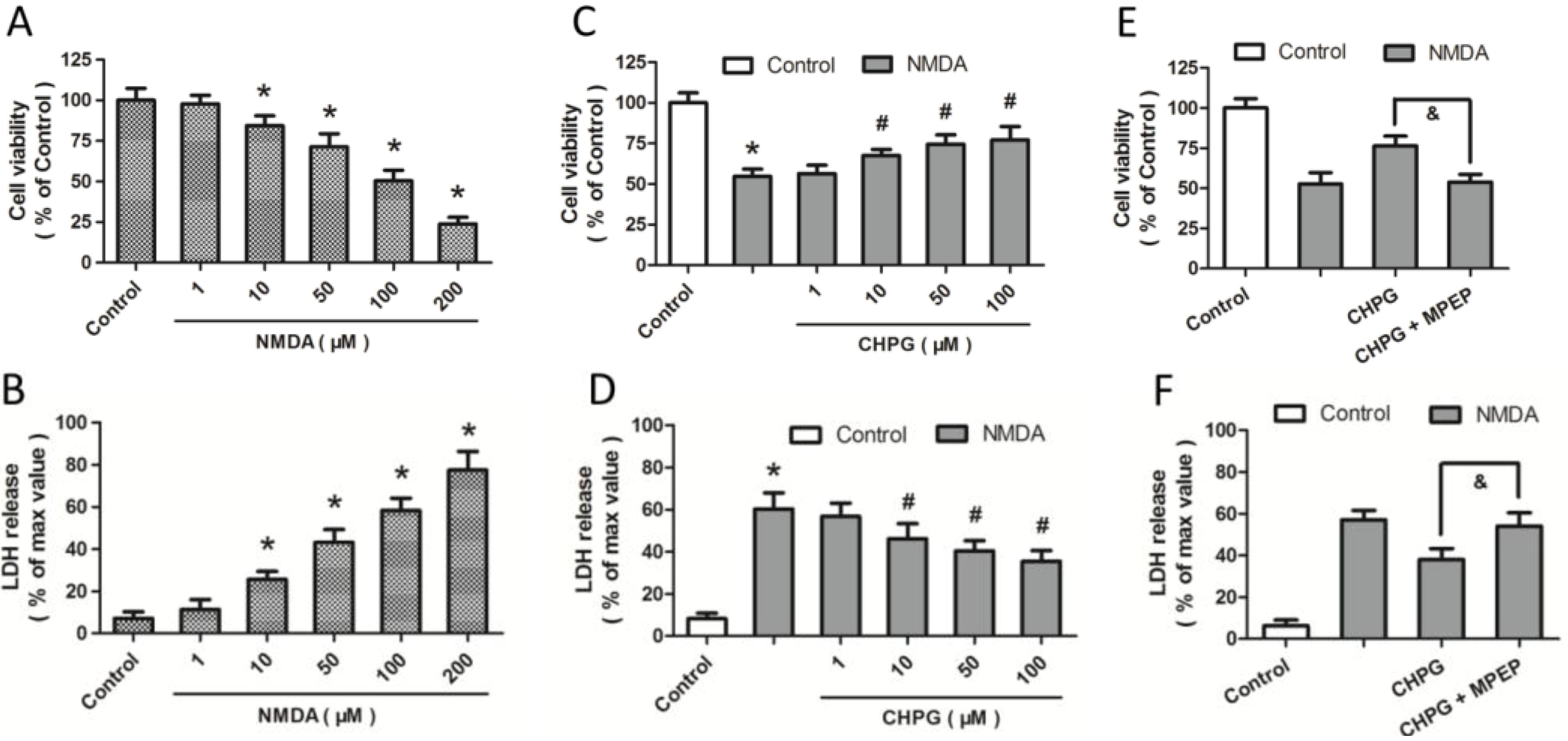
2.1. R,S-2-Chloro-5-hydroxyphenylglycine (CHPG) Protects against NMDA-Induced Neurotoxicity
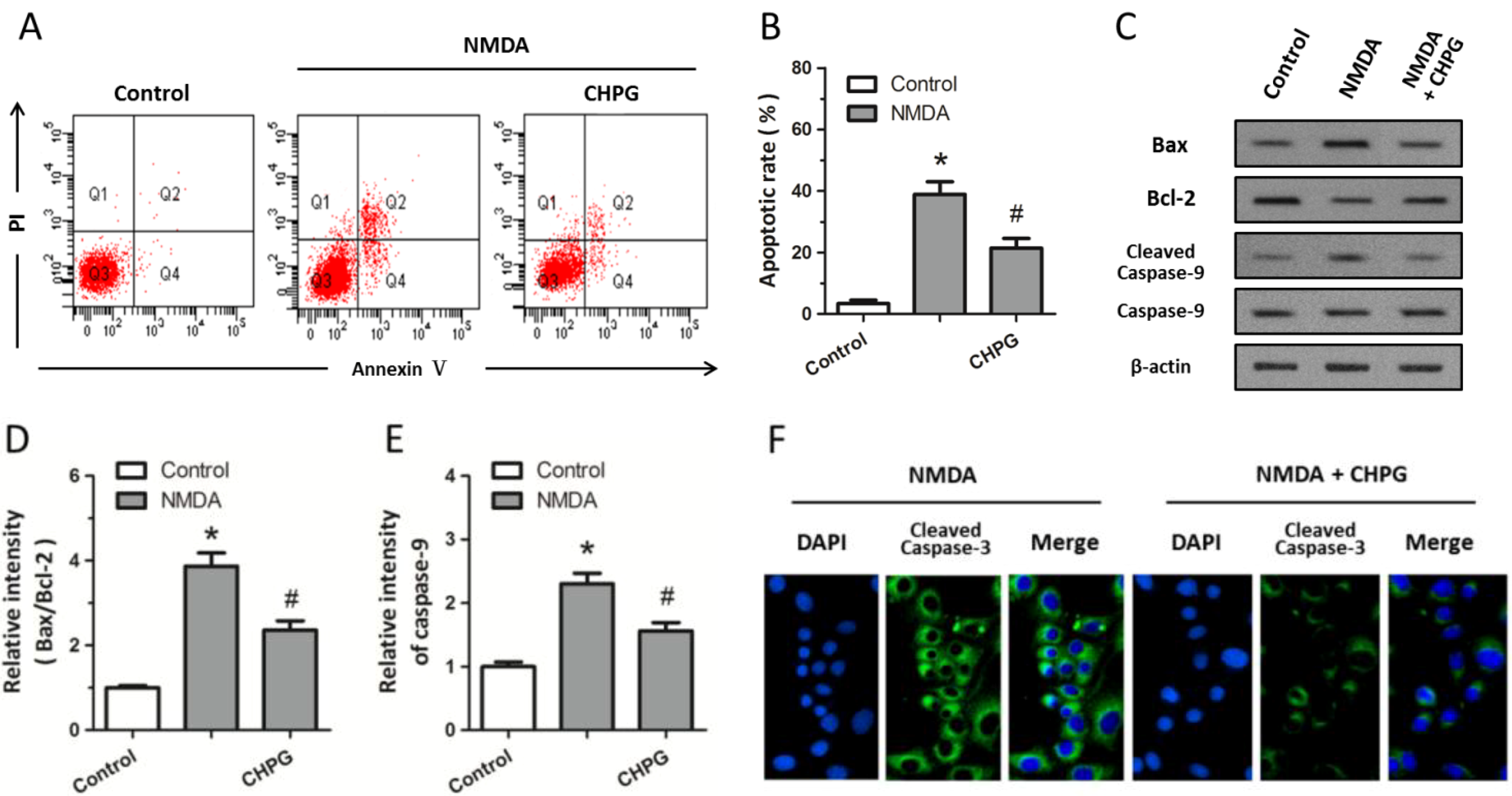
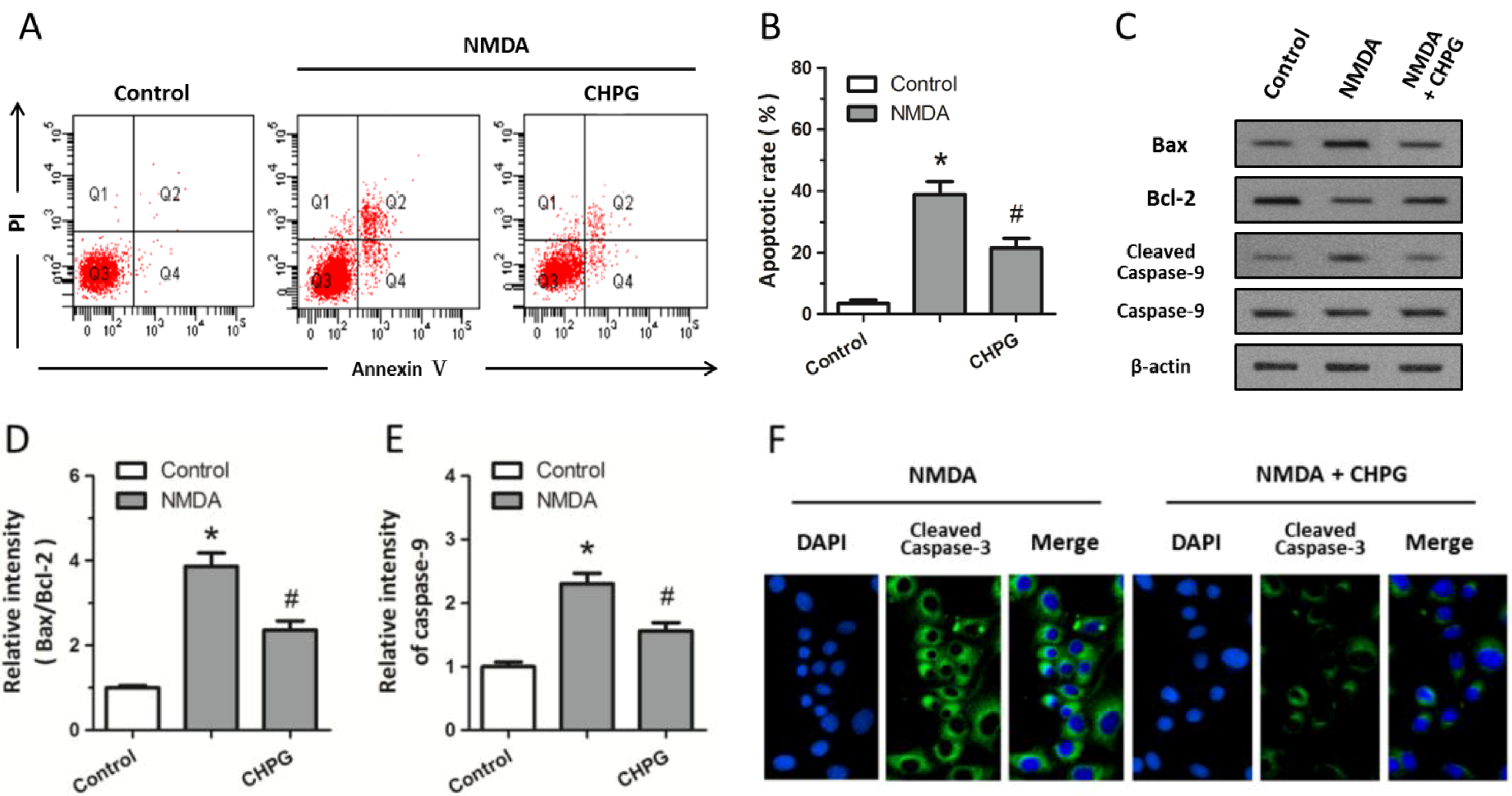
2.2. CHPG Inhibits NMDA-Induced Apoptotic Cell Death
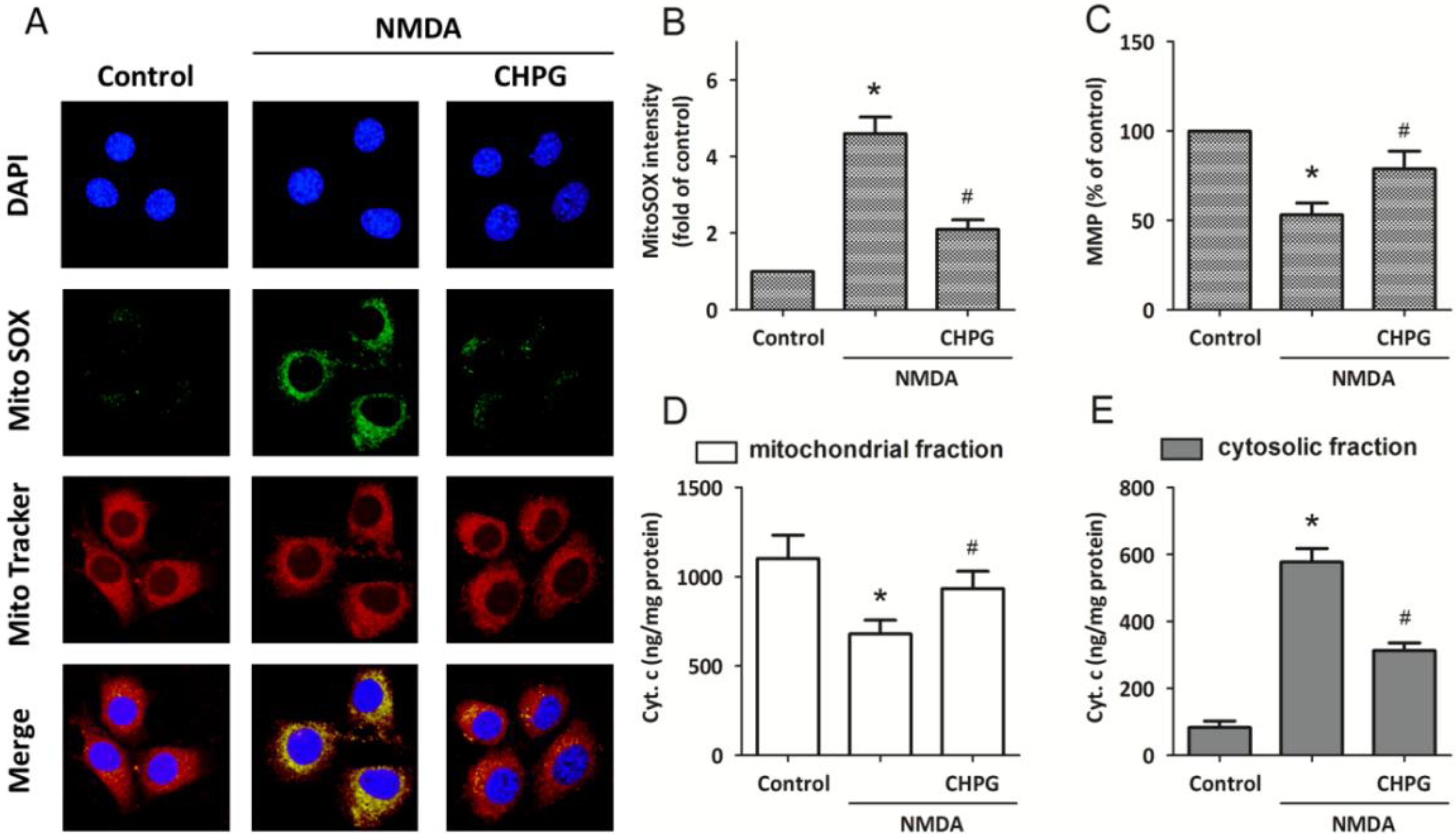
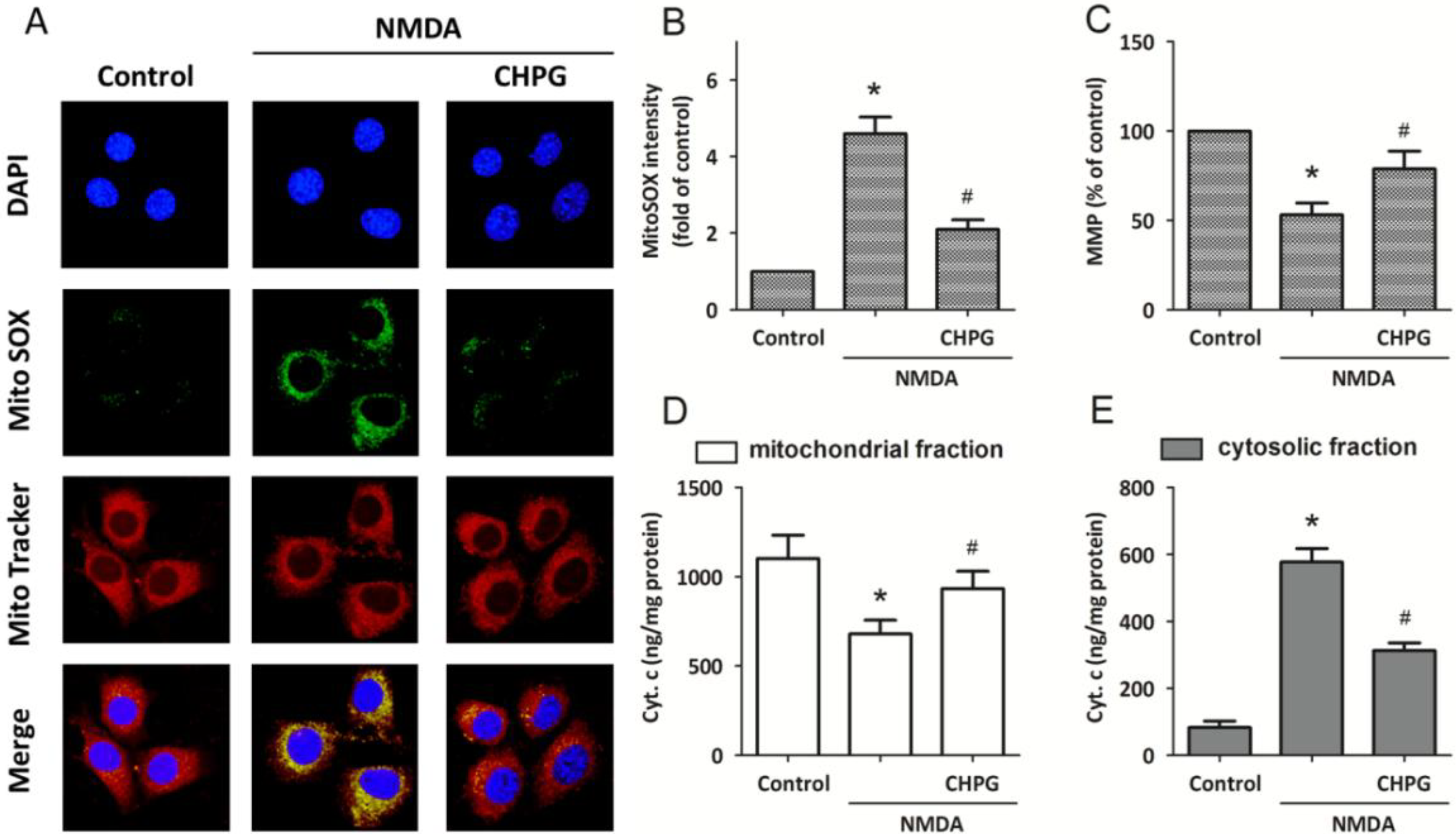
2.3. CHPG Attenuates NMDA-Induced Mitochondrial Dysfunction
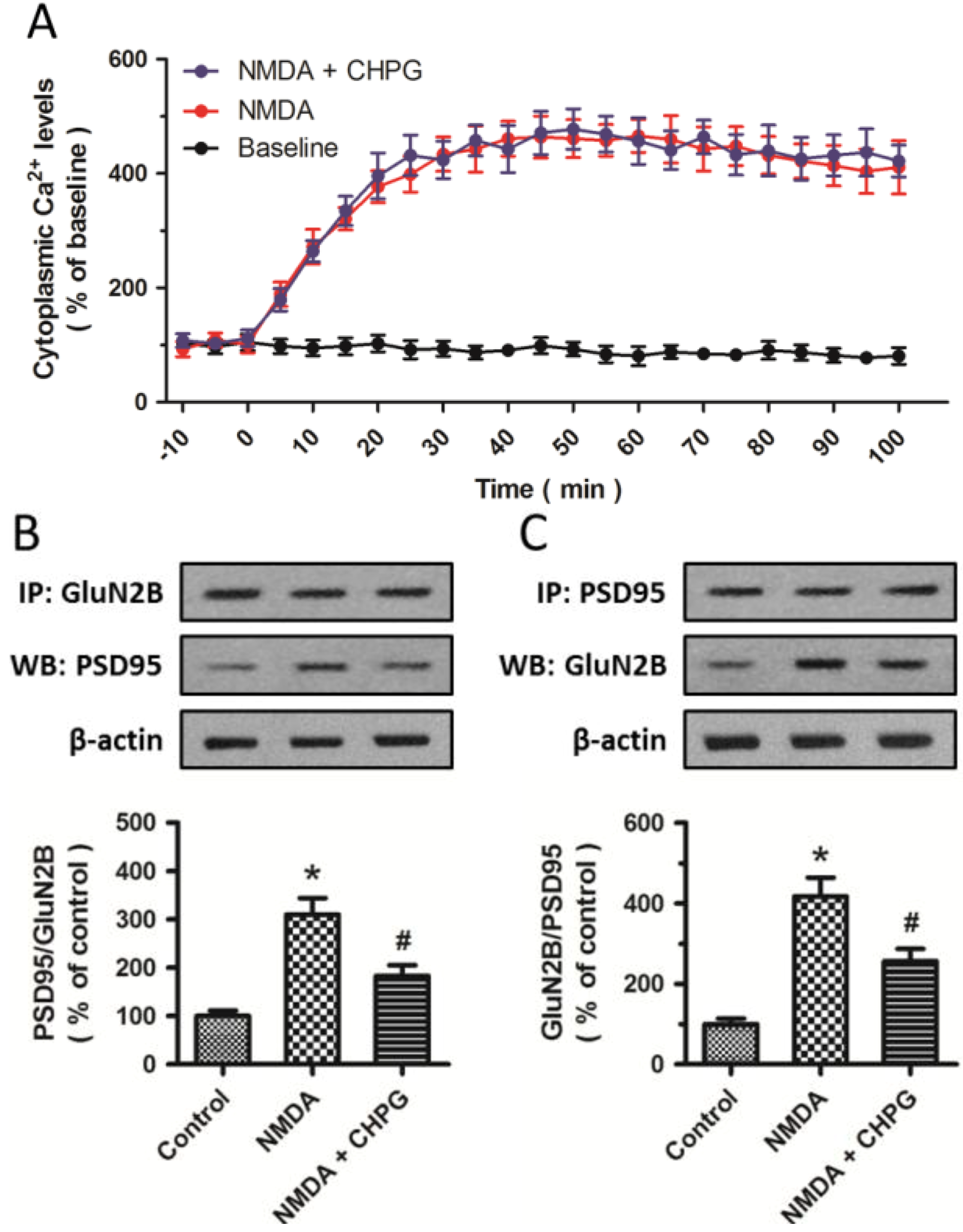
2.4. CHPG Disrupts the Formation of NMDAR-PSD-95 Complex

2.5. Discussion
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Cell Culture
3.3. Cell Viability Assay
3.4. LDH (Lactate Dehydrogenase) Release Assay
3.5. Flow Cytometry
3.6. Immunostaining
3.7. MitoSOX Assay
3.8. Measurement of Mitochondrial Membrane Potential (MMP)
3.9. Quantification of Cytochrome C Release
3.10. Calcium Imaging
3.11. Coimmunoprecipitation
3.12. Western Blotting
3.13. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Durand, D.; Pampillo, M.; Caruso, C.; Lasaga, M. Role of metabotropic glutamate receptors in the control of neuroendocrine function. Neuropharmacology 2008, 55, 577–583. [Google Scholar]
- Brittain, J.M.; Chen, L.; Wilson, S.M.; Brustovetsky, T.; Gao, X.; Ashpole, N.M.; Molosh, A.I.; You, H.; Hudmon, A.; Shekhar, A.; et al. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J. Biol. Chem. 2011, 286, 37778–37792. [Google Scholar]
- Brennan-Minnella, A.M.; Shen, Y.; El-Benna, J.; Swanson, R.A. Phosphoinositide 3-kinase couples NMDA receptors to superoxide release in excitotoxic neuronal death. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Degos, V.; Peineau, S.; Nijboer, C.; Kaindl, A.M.; Sigaut, S.; Favrais, G.; Plaisant, F.; Teissier, N.; Gouadon, E.; Lombet, A.; et al. G protein-coupled receptor kinase 2 and group I metabotropic glutamate receptors mediate inflammation-induced sensitization to excitotoxic neurodegeneration. Ann. Neurol. 2013, 73, 667–678. [Google Scholar]
- Hsieh, M.H.; Ho, S.C.; Yeh, K.Y.; Pawlak, C.R.; Chang, H.M.; Ho, Y.J.; Lai, T.J.; Wu, F.Y. Blockade of metabotropic glutamate receptors inhibits cognition and neurodegeneration in an MPTP-induced Parkinson’s disease rat model. Pharmacol. Biochem. Behav. 2012, 102, 64–71. [Google Scholar]
- Sheng, M. The postsynaptic NMDA-receptor—PSD-95 signaling complex in excitatory synapses of the brain. J. Cell Sci. 2001, 114, 1251. [Google Scholar]
- Martel, M.A.; Ryan, T.J.; Bell, K.F.; Fowler, J.H.; McMahon, A.; Al-Mubarak, B.; Komiyama, N.H.; Horsburgh, K.; Kind, P.C.; Grant, S.G.; et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron 2012, 74, 543–556. [Google Scholar]
- Li, B.; Otsu, Y.; Murphy, T.H.; Raymond, L.A. Developmental decrease in NMDA receptor desensitization associated with shift to synapse and interaction with postsynaptic density-95. J. Neurosci. 2003, 23, 11244–11254. [Google Scholar]
- Hu, S.Q.; Zhu, J.; Pei, D.S.; Zong, Y.Y.; Yan, J.Z.; Hou, X.Y.; Zhang, G.Y. Overexpression of the PDZ1 domain of PSD-95 diminishes ischemic brain injury via inhibition of the GluR6.PSD-95.MLK3 pathway. J. Neurosci. Res. 2009, 87, 3626–3638. [Google Scholar]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 2002, 298, 846–850. [Google Scholar]
- Jin, D.Z.; Guo, M.L.; Xue, B.; Mao, L.M.; Wang, J.Q. Differential regulation of CaMKIIα interactions with mGluR5 and NMDA receptors by Ca2+ in neurons. J. Neurochem. 2013, 127, 620–631. [Google Scholar]
- Alagarsamy, S.; Rouse, S.T.; Junge, C.; Hubert, G.W.; Gutman, D.; Smith, Y.; Conn, P.J. NMDA-induced phosphorylation and regulation of mGluR5. Pharmacol. Biochem. Behav. 2002, 73, 299–306. [Google Scholar]
- Matta, J.A.; Ashby, M.C.; Sanz-Clemente, A.; Roche, K.W.; Isaac, J.T. mGluR5 and NMDA receptors drive the experience- and activity-dependent NMDA receptor NR2B to NR2A subunit switch. Neuron 2011, 70, 339–351. [Google Scholar]
- Sureda, F.X.; Viu, E.; Zapata, A.; Capdevila, J.L.; Camins, A.; Escubedo, E.; Camarasa, J.; Trullas, R. Modulation of NMDA-induced cytosolic calcium levels by ACPC in cultured cerebellar granule cells. Neuroreport 1996, 7, 1824–1828. [Google Scholar]
- Chen, T.; Fei, F.; Jiang, X.F.; Zhang, L.; Qu, Y.; Huo, K.; Fei, Z. Down-regulation of Homer1b/c attenuates glutamate-mediated excitotoxicity through endoplasmic reticulum and mitochondria pathways in rat cortical neurons. Free Radic. Biol. Med. 2012, 52, 208–217. [Google Scholar]
- Byrnes, K.R.; Loane, D.J.; Stoica, B.A.; Zhang, J.; Faden, A.I. Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J. Neuroinflamm. 2012, 9. [Google Scholar] [CrossRef]
- Izumi, Y.; Zorumski, C.F. NMDA receptors, mGluR5, and endocannabinoids are involved in a cascade leading to hippocampal long-term depression. Neuropsychopharmacology 2012, 37, 609–617. [Google Scholar]
- Ben-Mabrouk, F.; Amos, L.B.; Tryba, A.K. Metabotropic glutamate receptors (mGluR5) activate transient receptor potential canonical channels to improve the regularity of the respiratory rhythm generated by the pre-Botzinger complex in mice. Eur. J. Neurosci. 2012, 35, 1725–1737. [Google Scholar]
- Gerber, U.; Gee, C.E.; Benquet, P. Metabotropic glutamate receptors: intracellular signaling pathways. Curr. Opin. Pharmacol. 2007, 7, 56–61. [Google Scholar]
- O’Leary, D.M.; Movsesyan, V.; Vicini, S.; Faden, A.I. Selective mGluR5 antagonists MPEP and SIB-1893 decrease NMDA or glutamate-mediated neuronal toxicity through actions that reflect NMDA receptor antagonism. Br. J. Pharmacol. 2000, 131, 1429–1437. [Google Scholar]
- Movsesyan, V.A.; O’Leary, D.M.; Fan, L.; Bao, W.; Mullins, P.G.; Knoblach, S.M.; Faden, A.I. mGluR5 antagonists 2-methyl-6-(phenylethynyl)-pyridine and (E)-2-methyl-6-(2-phenylethenyl)-pyridine reduce traumatic neuronal injury in vitro and in vivo by antagonizing N-methyl-d-aspartate receptors. J. Pharmacol. Exp. Ther. 2001, 296, 41–47. [Google Scholar]
- Allen, J.W.; Knoblach, S.M.; Faden, A.I. Activation of group I metabotropic glutamate receptors reduces neuronal apoptosis but increases necrotic cell death in vitro. Cell Death Differ. 2000, 7, 470–476. [Google Scholar]
- Movsesyan, V.A.; Stoica, B.A.; Faden, A.I. MGLuR5 activation reduces beta-amyloid-induced cell death in primary neuronal cultures and attenuates translocation of cytochrome c and apoptosis-inducing factor. J. Neurochem. 2004, 89, 1528–1536. [Google Scholar]
- Kemp, A.; Manahan-Vaughan, D. Hippocampal long-term depression and long-term potentiation encode different aspects of novelty acquisition. Proc. Natl. Acad. Sci. USA 2004, 101, 8192–8197. [Google Scholar]
- Chen, T.; Zhang, L.; Qu, Y.; Huo, K.; Jiang, X.; Fei, Z. The selective mGluR5 agonist CHPG protects against traumatic brain injury in vitro and in vivo via ERK and Akt pathway. Int. J. Mol. Med. 2012, 29, 630–636. [Google Scholar]
- Chen, T.; Cao, L.; Dong, W.; Luo, P.; Liu, W.; Qu, Y.; Fei, Z. Protective effects of mGluR5 positive modulators against traumatic neuronal injury through PKC-dependent activation of MEK/ERK pathway. Neurochem. Res. 2012, 37, 983–990. [Google Scholar]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar]
- Luo, P.; Yang, Y.; Liu, W.; Rao, W.; Bian, H.; Li, X.; Chen, T.; Liu, M.; Zhao, Y.; Dai, S.; et al. Downregulation of postsynaptic density-95-interacting regulator of spine morphogenesis reduces glutamate-induced excitotoxicity by differentially regulating glutamate receptors in rat cortical neurons. FEBS J. 2013, 280, 6114–6127. [Google Scholar]
- Meng, F.J.; Guo, J.; Song, B.; Yan, X.B.; Zhang, G.Y. Competitive binding of postsynaptic density 95 and Ca2+-calmodulin dependent protein kinase II to N-methyl-d-aspartate receptor subunit 2B in rat brain. Acta Pharmacol. Sin. 2004, 25, 176–180. [Google Scholar]
- Migaud, M.; Charlesworth, P.; Dempster, M.; Webster, L.C.; Watabe, A.M.; Makhinson, M.; He, Y.; Ramsay, M.F.; Morris, R.G.; Morrison, J.H.; et al. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 1998, 396, 433–439. [Google Scholar]
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 1999, 284, 1845–1848. [Google Scholar]
- Zhou, L.; Li, F.; Xu, H.B.; Luo, C.X.; Wu, H.Y.; Zhu, M.M.; Lu, W.; Ji, X.; Zhou, Q.G.; Zhu, D.Y. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat. Med. 2010, 16, 1439–1443. [Google Scholar]
- Zieminska, E.; Stafiej, A.; Lazarewicz, J.W. Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurones. Neurochem. Int. 2003, 43, 481–492. [Google Scholar]
- Blaabjerg, M.; Kristensen, B.W.; Bonde, C.; Zimmer, J. The metabotropic glutamate receptor agonist 1S,3R-ACPD stimulates and modulates NMDA receptor mediated excitotoxicity in organotypic hippocampal slice cultures. Brain Res. 2001, 898, 91–104. [Google Scholar]
- Bonsi, P.; Platania, P.; Martella, G.; Madeo, G.; Vita, D.; Tassone, A.; Bernardi, G.; Pisani, A. Distinct roles of group I mGlu receptors in striatal function. Neuropharmacology 2008, 55, 392–395. [Google Scholar]
- Huang, H.; van den Pol, A.N. Rapid direct excitation and long-lasting enhancement of NMDA response by group I metabotropic glutamate receptor activation of hypothalamic melanin-concentrating hormone neurons. J. Neurosci. 2007, 27, 11560–11572. [Google Scholar]
- Ciruela, F.; Soloviev, M.M.; Chan, W.Y.; McIlhinney, R.A. Homer-1c/Vesl-1L modulates the cell surface targeting of metabotropic glutamate receptor type 1α: Evidence for an floating function. Mol. Cell. Neurosci. 2000, 15, 36–50. [Google Scholar]
- Padanyi, R.; Paszty, K.; Strehler, E.E.; Enyedi, A. PSD-95 mediates membrane clustering of the human plasma membrane Ca2+ pump isoform 4b. Biochim. Biophys. Acta 2009, 1793, 1023–1032. [Google Scholar]
- Hayashi, M.K.; Tang, C.; Verpelli, C.; Narayanan, R.; Stearns, M.H.; Xu, R.M.; Li, H.; Sala, C.; Hayashi, Y. The postsynaptic density proteins Homer and Shank form a polymeric network structure. Cell 2009, 137, 159–171. [Google Scholar]
- Khoboko, T.; Russell, V.A. Effects of development and dopamine depletion on striatal NMDA receptor-mediated calcium uptake. Metab. Brain Dis. 2008, 23, 9–30. [Google Scholar]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar]
- Chen, T.; Yang, Y.F.; Luo, P.; Liu, W.; Dai, S.H.; Zheng, X.R.; Fei, Z.; Jiang, X.F. Homer1 knockdown protects dopamine neurons through regulating calcium homeostasis in an in vitro model of Parkinson’s disease. Cell Signal. 2013, 25, 2863–2870. [Google Scholar]
- Chen, T.; Zhu, J.; Zhang, C.; Huo, K.; Fei, Z.; Jiang, X.F. Protective effects of SKF-96365, a non-specific inhibitor of SOCE, against MPP+-induced cytotoxicity in PC12 cells: Potential role of Homerl. PLoS One 2013, 8, e55601. [Google Scholar]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar]
- Friberg, H.; Wieloch, T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie 2002, 84, 241–250. [Google Scholar]
- Norenberg, M.D.; Rao, K.V. The mitochondrial permeability transition in neurologic disease. Neurochem. Int. 2007, 50, 983–997. [Google Scholar]
- Gibson, G.E.; Starkov, A.; Blass, J.P.; Ratan, R.R.; Beal, M.F. Cause and consequence: Mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 122–134. [Google Scholar]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar]
- Lipton, S.A. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 2004, 1, 101–110. [Google Scholar]
- Wang, P.; Zhang, H.; Chu, F.; Xu, X.; Lin, J.; Chen, C.; Li, G.; Cheng, Y.; Wang, L.; Li, Q.; et al. Synthesis and protective effect of new ligustrazine-benzoic acid derivatives against CoCl2-induced neurotoxicity in differentiated PC12 cells. Molecules 2013, 18, 13027–13042. [Google Scholar]
- Sung, D.K.; Chang, Y.S.; Kang, S.; Song, H.Y.; Park, W.S.; Lee, B.H. Comparative evaluation of hypoxic-ischemic brain injury by flow cytometric analysis of mitochondrial membrane potential with JC-1 in neonatal rats. J. Neurosci. Methods 2010, 193, 232–238. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dai, S.-H.; Qin, N.; Chen, T.; Luo, P.; Zhang, L.; Rao, W.; Yang, Y.-F.; Jiang, X.-F.; Fei, Z. Activation of mGluR5 Attenuates NMDA-Induced Neurotoxicity through Disruption of the NMDAR-PSD-95 Complex and Preservation of Mitochondrial Function in Differentiated PC12 Cells. Int. J. Mol. Sci. 2014, 15, 10892-10907. https://doi.org/10.3390/ijms150610892
Dai S-H, Qin N, Chen T, Luo P, Zhang L, Rao W, Yang Y-F, Jiang X-F, Fei Z. Activation of mGluR5 Attenuates NMDA-Induced Neurotoxicity through Disruption of the NMDAR-PSD-95 Complex and Preservation of Mitochondrial Function in Differentiated PC12 Cells. International Journal of Molecular Sciences. 2014; 15(6):10892-10907. https://doi.org/10.3390/ijms150610892
Chicago/Turabian StyleDai, Shu-Hui, Na Qin, Tao Chen, Peng Luo, Lei Zhang, Wei Rao, Yue-Fan Yang, Xiao-Fan Jiang, and Zhou Fei. 2014. "Activation of mGluR5 Attenuates NMDA-Induced Neurotoxicity through Disruption of the NMDAR-PSD-95 Complex and Preservation of Mitochondrial Function in Differentiated PC12 Cells" International Journal of Molecular Sciences 15, no. 6: 10892-10907. https://doi.org/10.3390/ijms150610892