The Effects of CoCl2 on HIF-1α Protein under Experimental Conditions of Autoprogressive Hypoxia Using Mouse Models

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
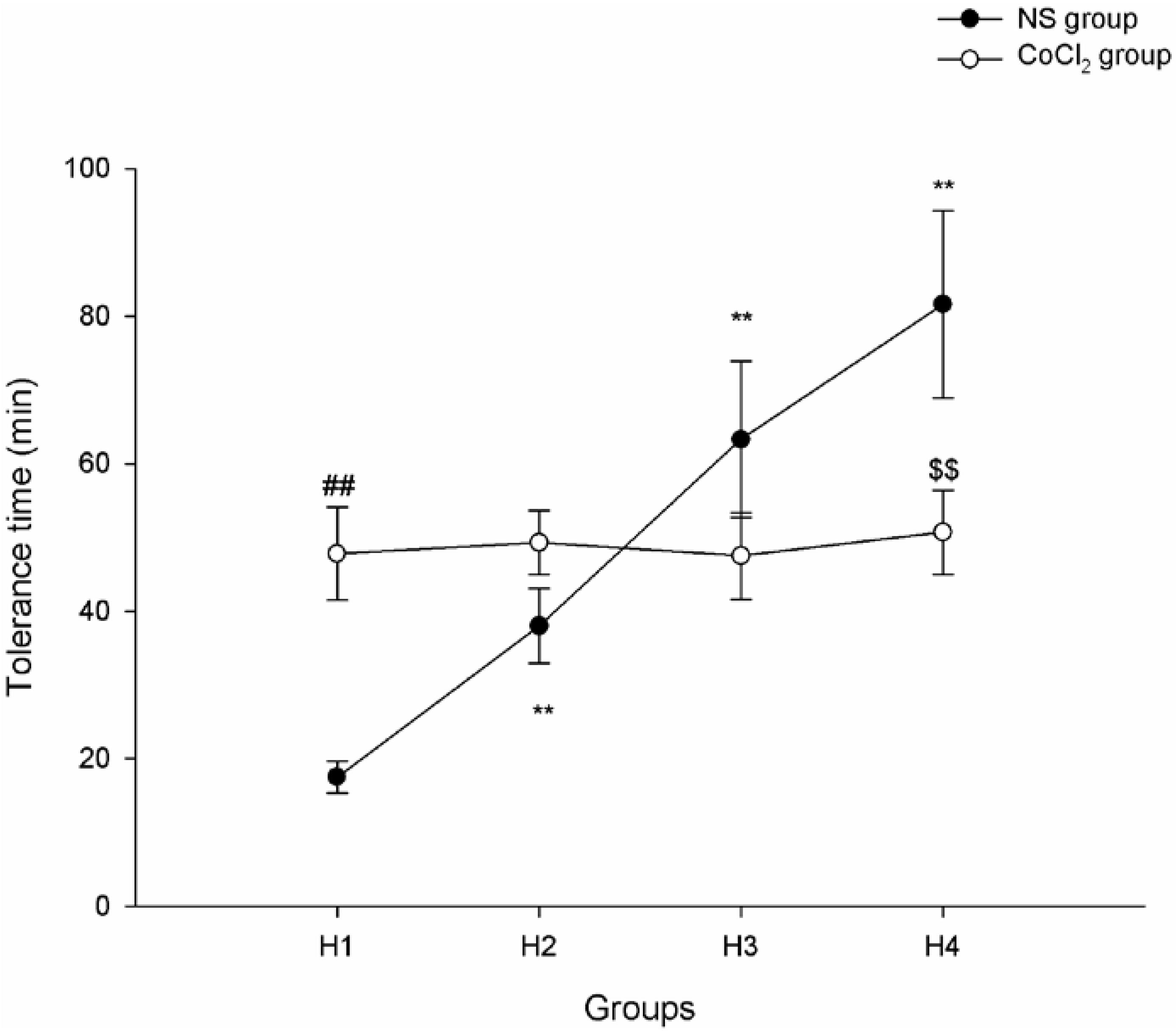
2.1. The Effect of CoCl2 Pretreatment on Hypoxia Tolerance
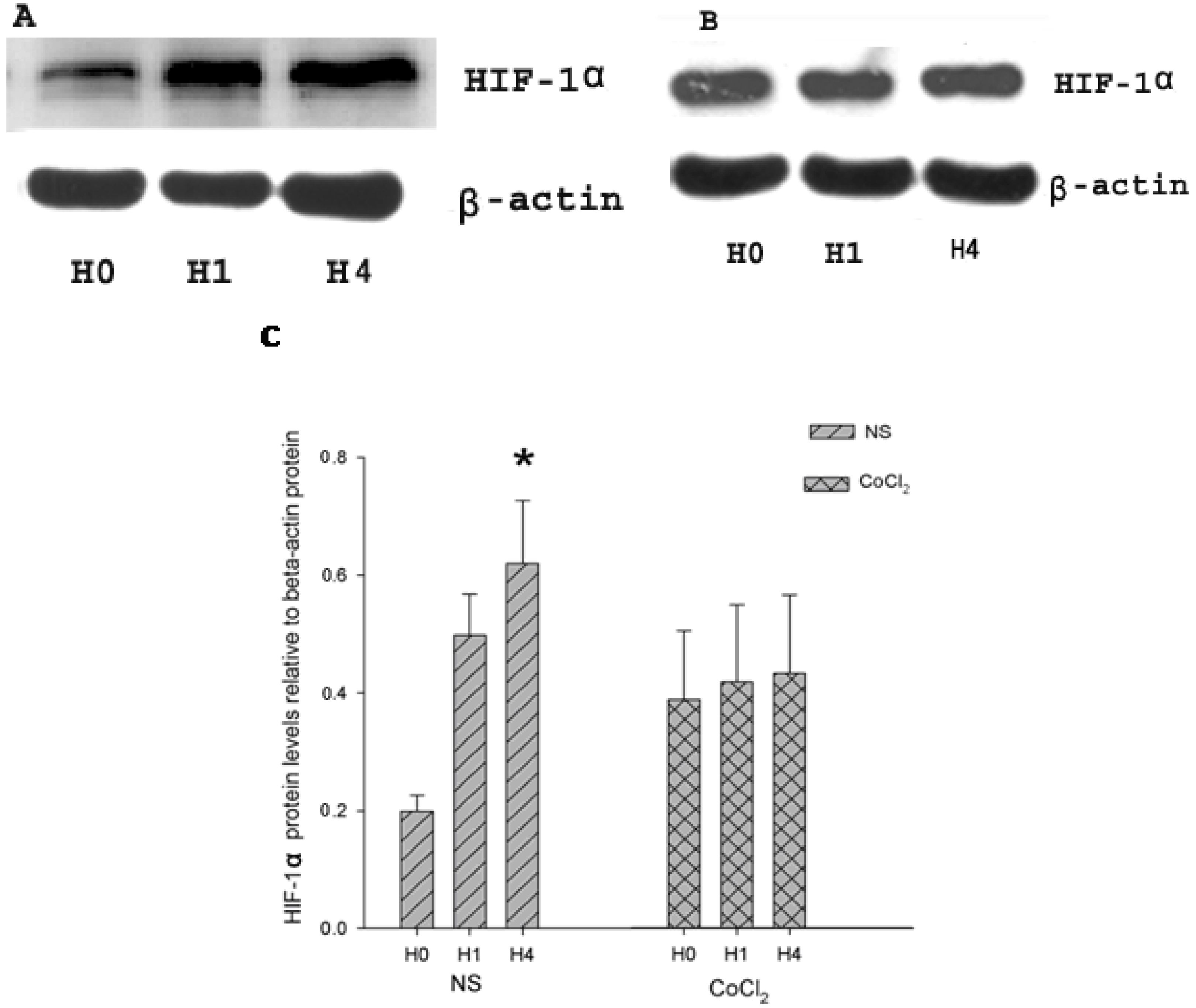
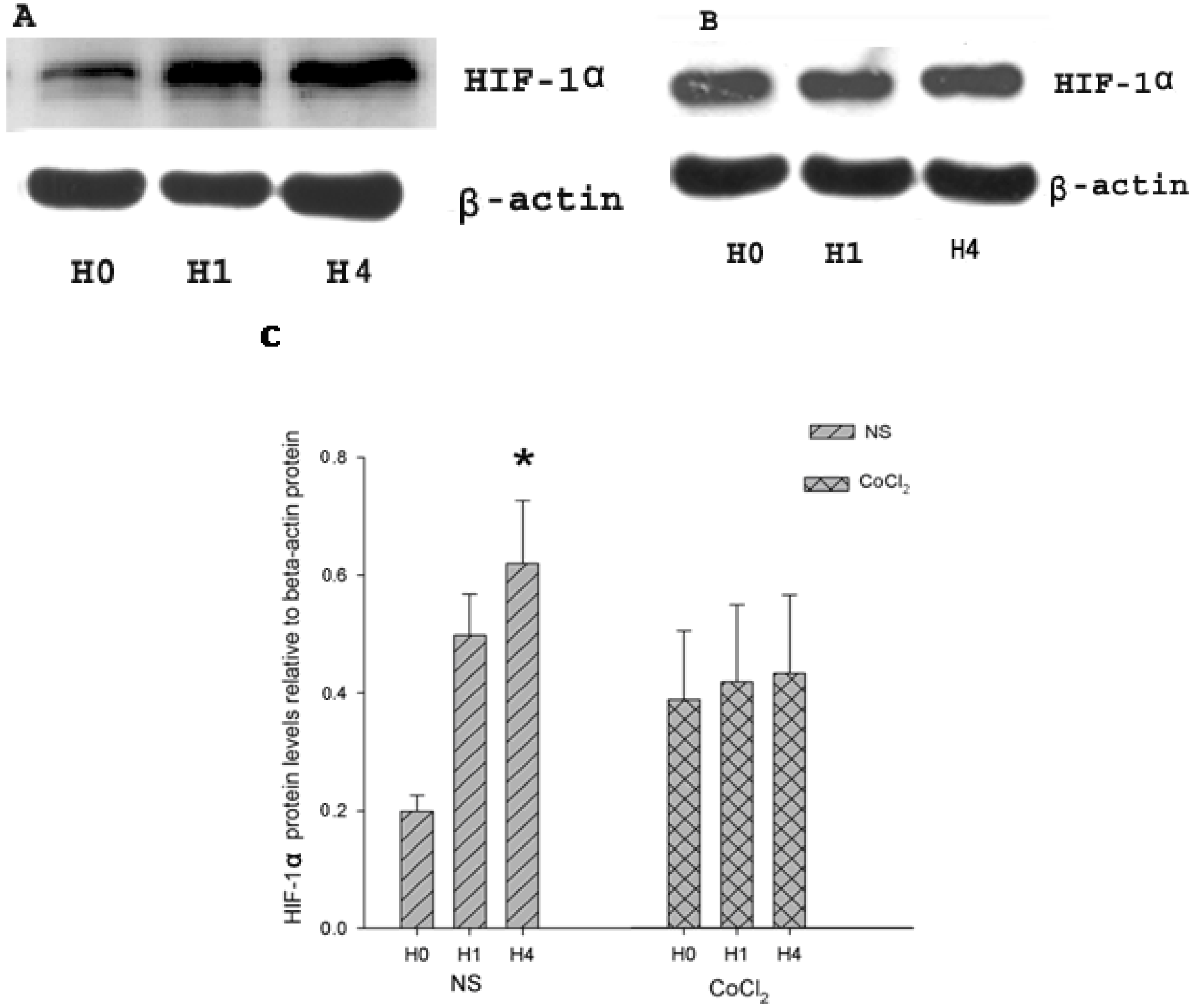
2.2. The Effect of CoCl2 Pretreatment on Hypoxia-Inducible Factor (HIF)-1α Protein Level
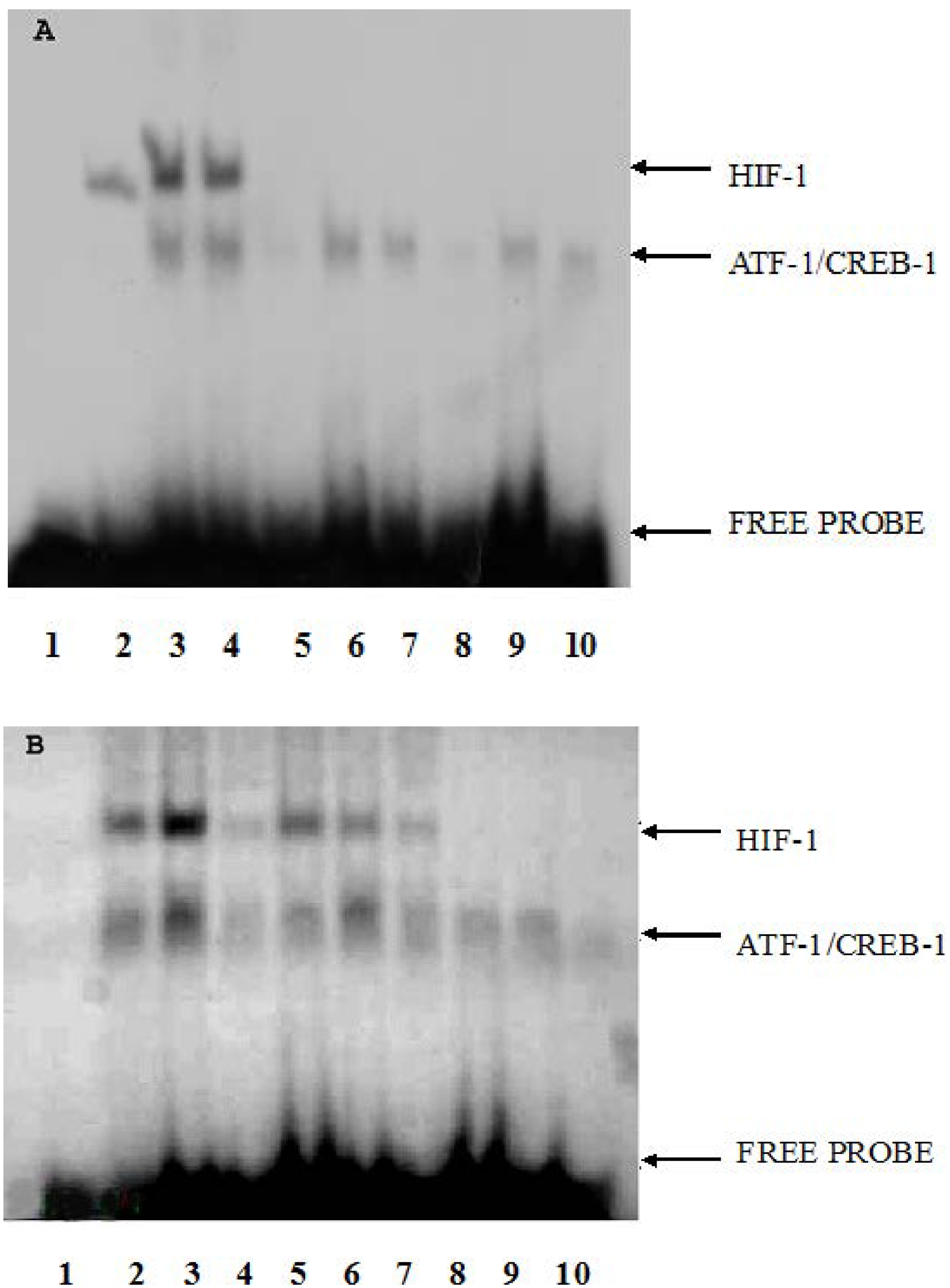
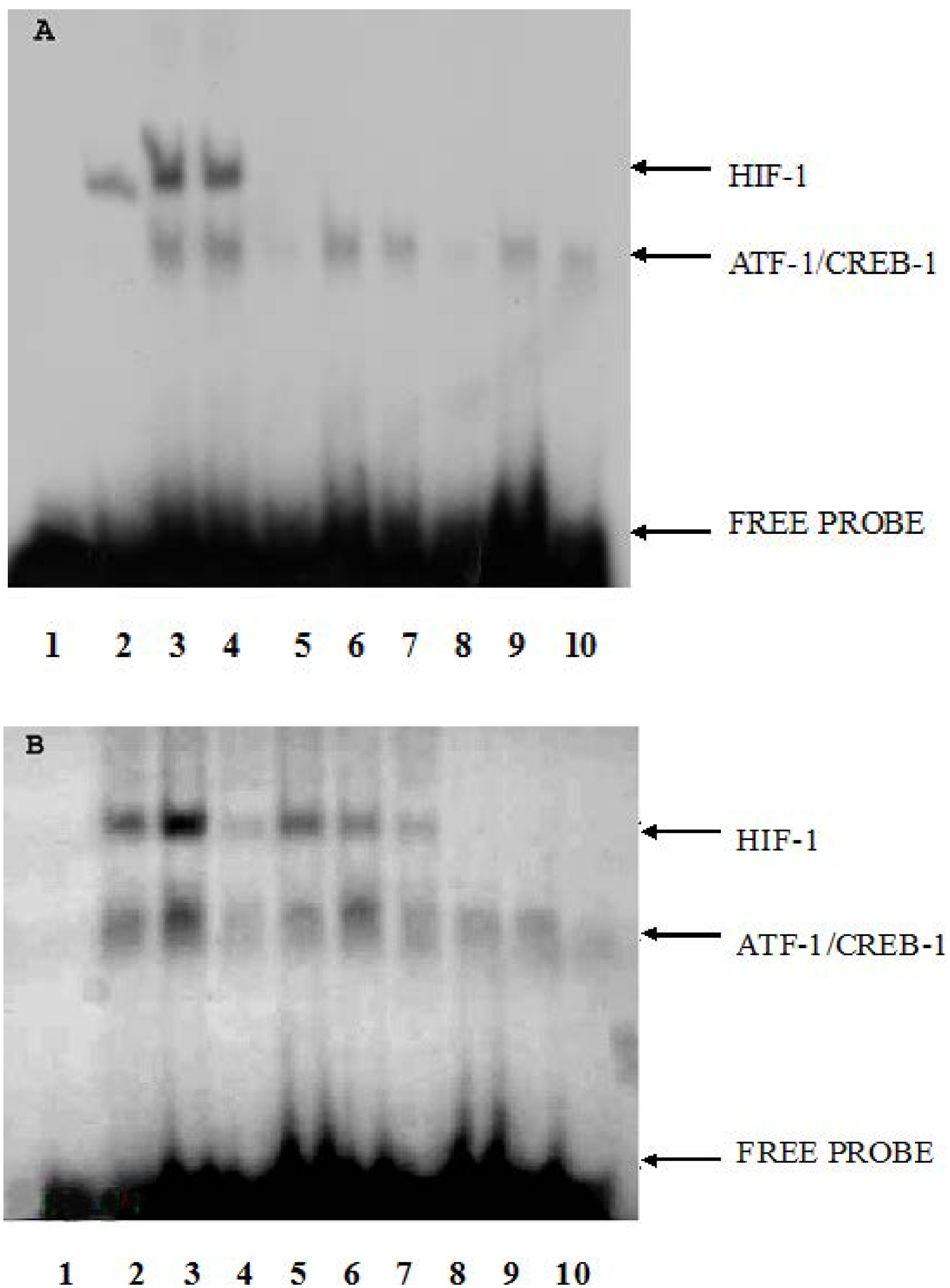
2.3. The Effect of CoCl2 Pretreatment on HIF-1 DNA Binding Activity
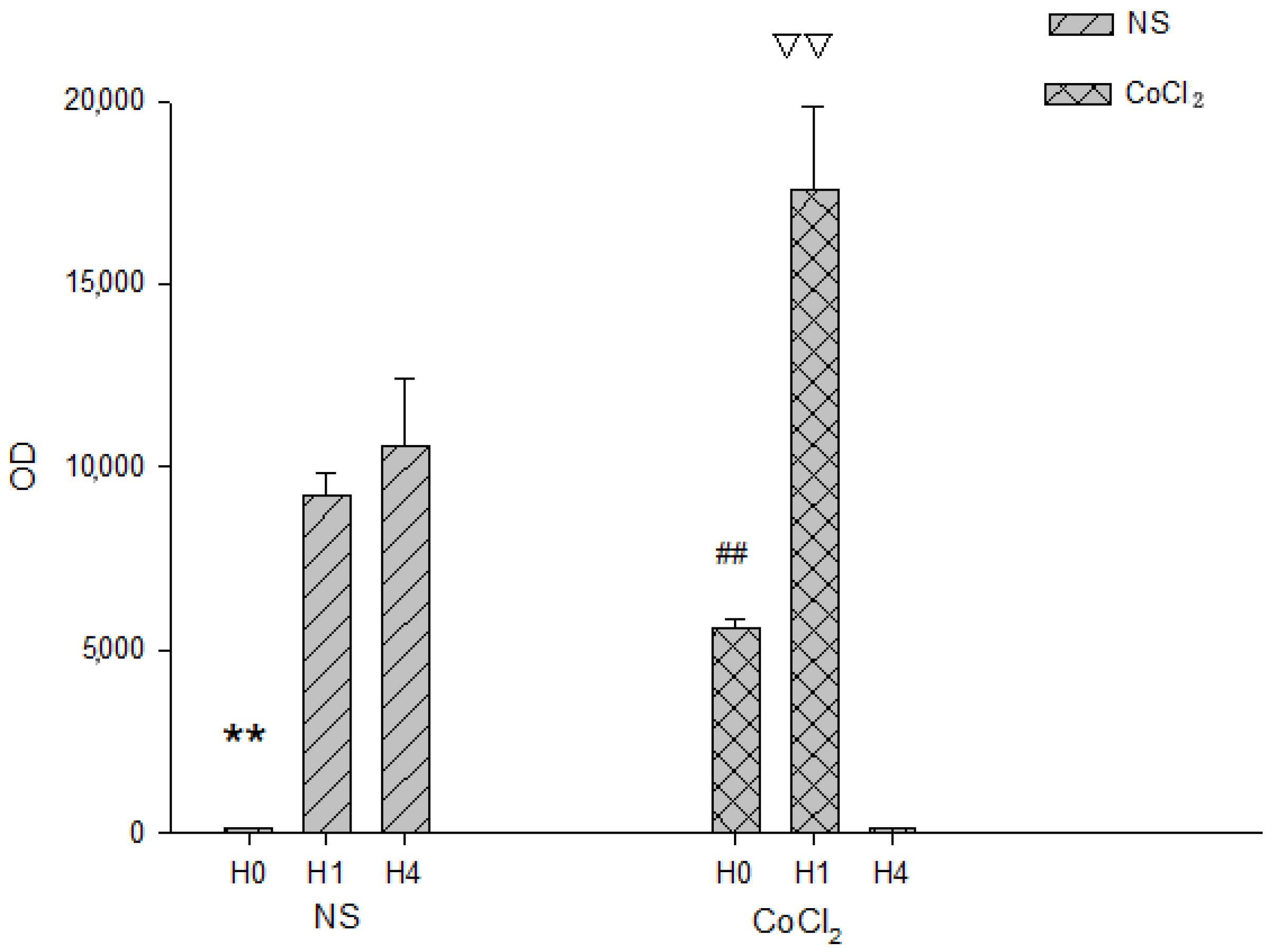
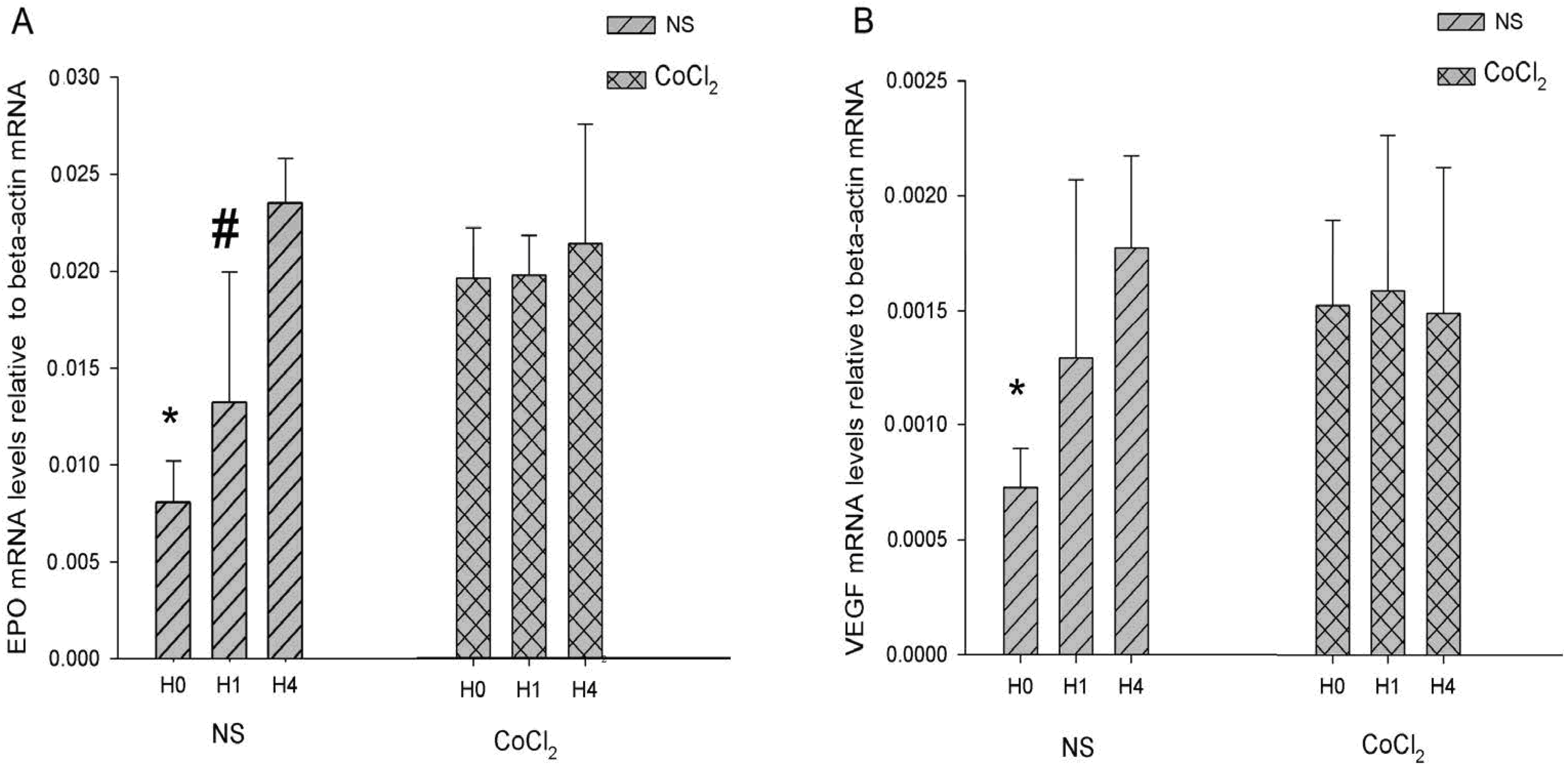
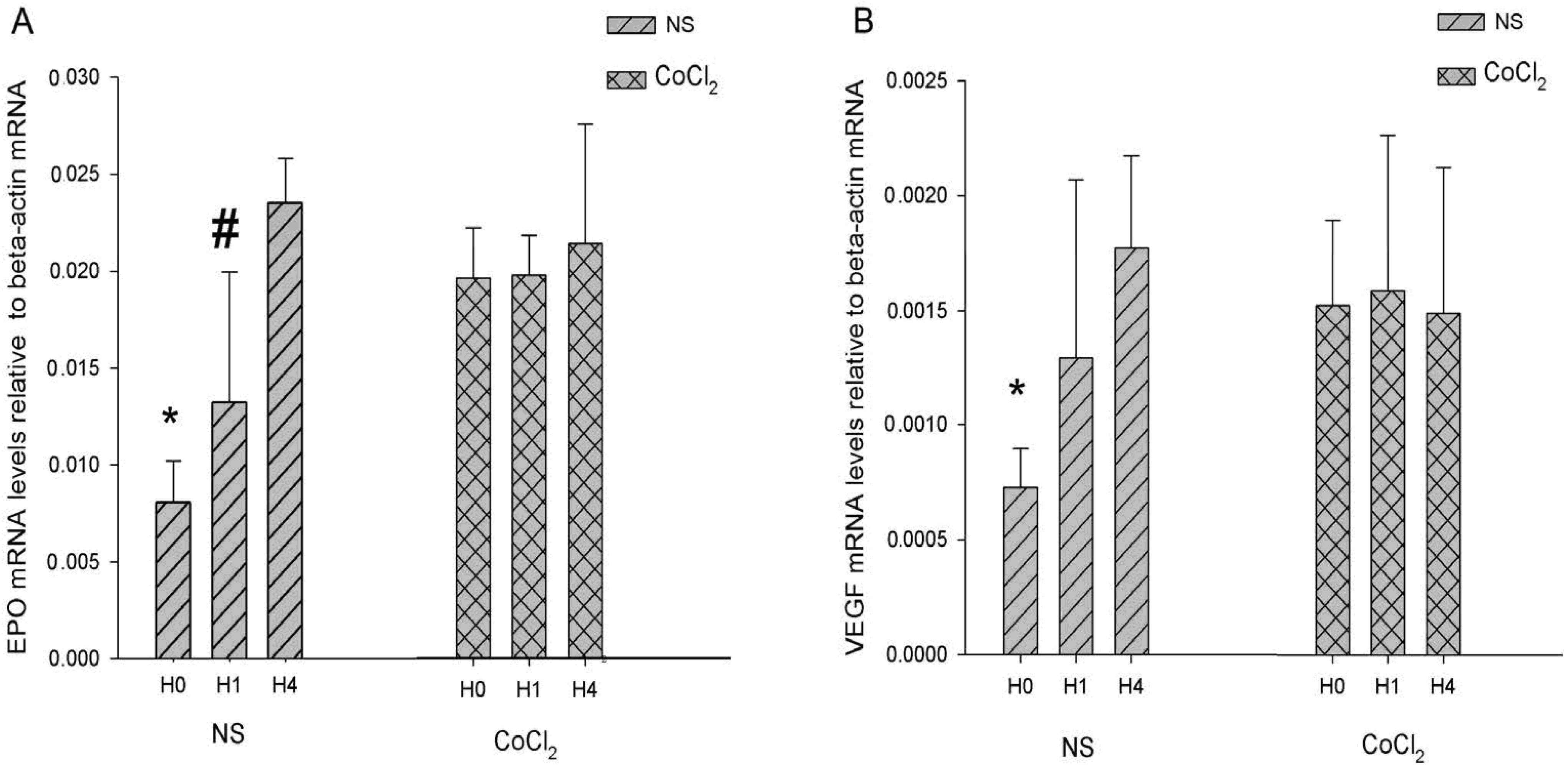
2.4. The Effect of CoCl2 Pretreatment on the Erythropoietin (EPO) and Vascular Endothelial Growth Factor (VEGF) mRNA Levels
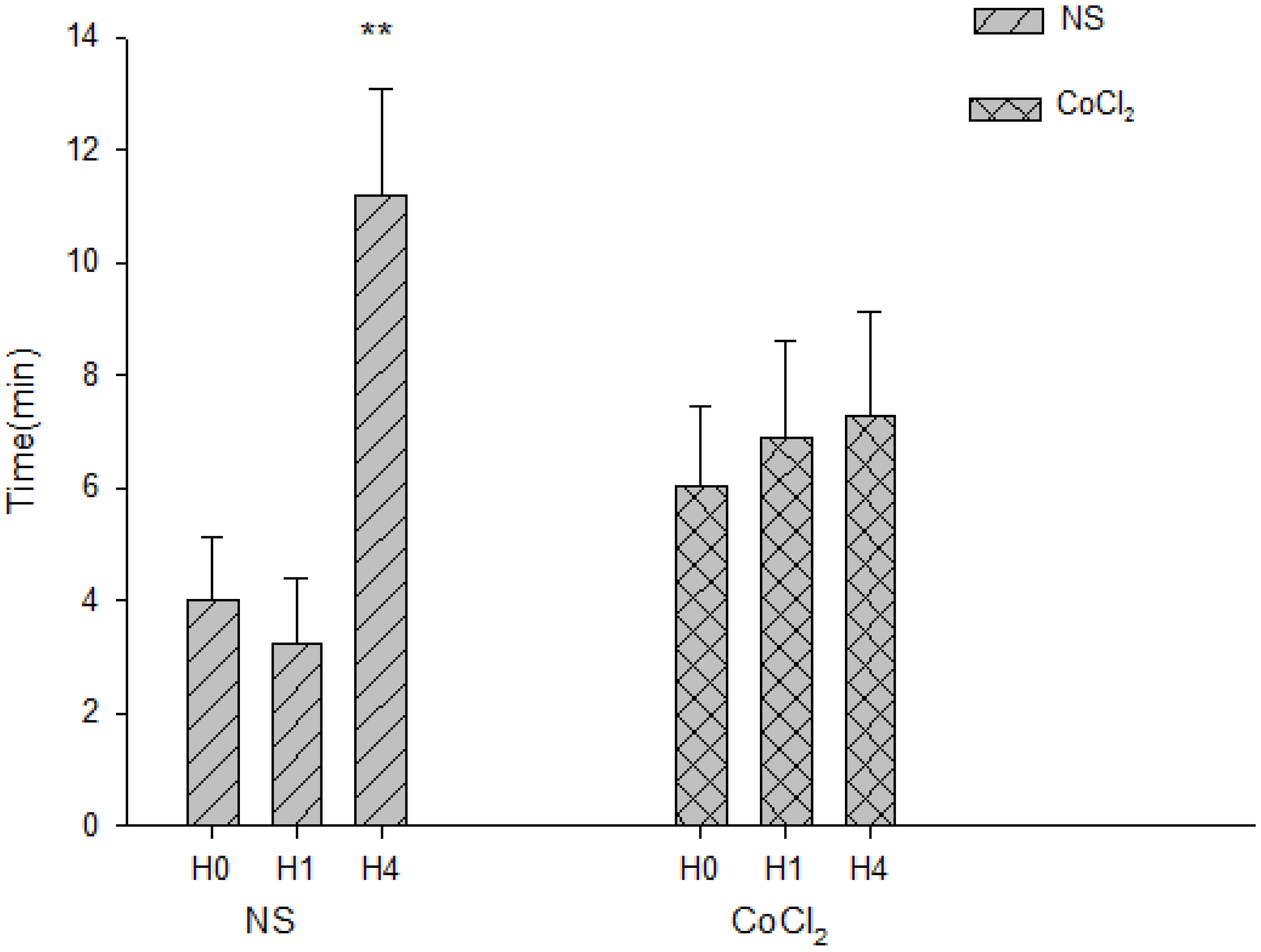
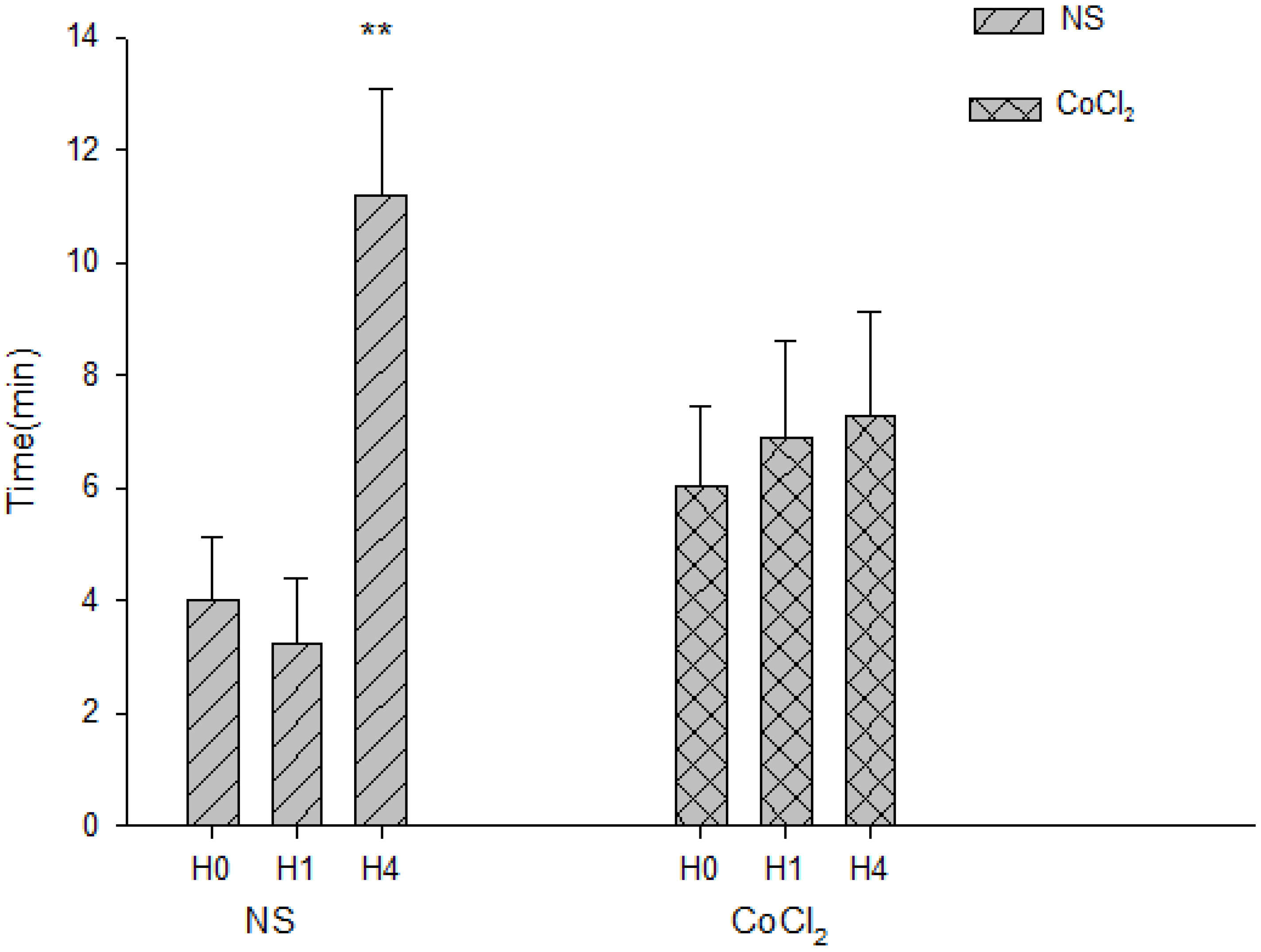
2.5. The Effect of CoCl2 Pretreatment on the Abolishment Time of Population Spikes (PS)
3. Experimental Section
3.1. Animal Model
3.2. Western Blot Analysis
3.3. Electrophoretic Mobility Shift Assay (EMSA)
3.4. Hippocampal Slice Preparation and PS Recording
3.5. RNA Isolation, cDNA Synthesis, and Real-Time PCR
3.6. Quantification and Statistical Analysis
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Kitagawa, K.; Matsumoto, M.; Tagaya, M.; Hata, R.; Ueda, H.; Niinobe, M.; Handa, N.; Fukunaga, R.; Kimura, K.; Mikoshiba, K.; et al. “Ischemic tolerance” phenomenon found in the brain. Brain Res. 1990, 528, 21–24. [Google Scholar] [CrossRef]
- Youssef, F.F.; Addae, J.I.; Stone, T.W. NMDA-induced preconditioning attenuates synaptic plasticity in the rat hippocampus. Brain Res. 2006, 1073–1074, 183–189. [Google Scholar] [CrossRef]
- Hoshi, A.; Yamamoto, T.; Shimizu, K.; Sugiura, Y.; Ugawa, Y. Chemical preconditioning-induced reactive astrocytosis contributes to the reduction of post-ischemic edema through aquaporin-4 down-regulation. Exp. Neurol. 2011, 227, 89–95. [Google Scholar] [CrossRef]
- Lu, G. Tissue-cell adaptation to hypoxia. Adv. Pathophysiol. 1963, 1, 197–239. [Google Scholar]
- Shao, G.; Lu, G.W. Hypoxic preconditioning in an autohypoxic animal model. Neurosci. Bull. 2012, 28, 316–320. [Google Scholar] [CrossRef]
- Shao, G.; Gao, C.Y.; Lu, G.W. Alterations of hypoxia-inducible factor-1 alpha in the hippocampus of mice acutely and repeatedly exposed to hypoxia. Neurosignals 2005, 14, 255–261. [Google Scholar]
- Shao, G.; Gong, K.R.; Li, J.; Xu, X.J.; Gao, C.Y.; Zeng, X.Z.; Lu, G.W.; Huo, X. Antihypoxic effects of neuroglobin in hypoxia-preconditioned mice and sh-sy5y cells. Neurosignals 2009, 17, 196–202. [Google Scholar] [CrossRef]
- Shao, G.; Zhang, R.; Wang, Z.L.; Gao, C.Y.; Huo, X.; Lu, G.W. Hypoxic preconditioning improves spatial cognitive ability in mice. Neurosignals 2006, 15, 314–321. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 inhibitors for cancer therapy: From gene expression to drug discovery. Curr. Pharm. Des. 2009, 15, 3839–3843. [Google Scholar] [CrossRef]
- Formenti, F.; Constantin-Teodosiu, D.; Emmanuel, Y.; Cheeseman, J.; Dorrington, K.L.; Edwards, L.M.; Humphreys, S.M.; Lappin, T.R.; McMullin, M.F.; McNamara, C.J.; et al. Regulation of human metabolism by hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2010, 107, 12722–12727. [Google Scholar] [CrossRef]
- Foley, R.N. Emerging erythropoiesis-stimulating agents. Nat. Rev. Nephrol. 2010, 6, 218–223. [Google Scholar] [CrossRef]
- Yu, B.; Miao, Z.H.; Jiang, Y.; Li, M.H.; Yang, N.; Li, T.; Ding, J. C-jun protects hypoxia-induciblefactor-1alpha from degradation via its oxygen-dependent degradation domain in a nontranscriptional manner. Cancer Res. 2009, 69, 7704–7712. [Google Scholar] [CrossRef]
- Yuan, Y.; Hilliard, G.; Ferguson, T.; Millhorn, D.E. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von hippel-lindau protein by direct binding to hypoxia-inducible factor-alpha. J. Biol. Chem. 2003, 278, 15911–15916. [Google Scholar] [CrossRef]
- Samelko, L.; Caicedo, M.S.; Lim, S.J.; Della-Valle, C.; Jacobs, J.; Hallab, N.J. Cobalt-alloy implant debris induce HIF-1α hypoxia associated responses: A mechanism for metal-specific orthopedic implant failure. PLoS One 2013, 8, e67127. [Google Scholar]
- Shrivastava, K.; Ram, M.S.; Bansal, A.; Singh, S.S.; Ilavazhagan, G. Cobalt supplementation promotes hypoxic tolerance and facilitates acclimatization to hypobaric hypoxia in rat brain. High Alt. Med. Biol. 2008, 9, 63–75. [Google Scholar] [CrossRef]
- Shrivastava, K.; Shukla, D.; Bansal, A.; Sairam, M.; Banerjee, P.K.; Ilavazhagan, G. Neuroprotective effect of cobalt chloride on hypobaric hypoxia-induced oxidative stress. Neurochem. Int. 2008, 52, 368–375. [Google Scholar] [CrossRef]
- Bae, S.; Jeong, H.J.; Cha, H.J.; Kim, K.; Choi, Y.M.; An, I.S.; Koh, H.J.; Lim, D.J.; Lee, S.J.; An, S. The hypoxia-mimetic agent cobalt chloride induces cell cycle arrest and alters gene expression in u266 multiple myeloma cells. Int. J. Mol. Med. 2012, 30, 1180–1186. [Google Scholar]
- Oh, S.W.; Ahn, J.M.; Lee, Y.M.; Kim, S.; Chin, H.J.; Chae, D.W.; Na, K.Y. Activation of hypoxia-inducible factor by cobalt is associated with the attenuation of tissue injury and apoptosis in cyclosporine-induced nephropathy. Tohoku J. Exp. Med. 2012, 226, 197–206. [Google Scholar] [CrossRef]
- Lu, G.W.; Yu, S.; Li, R.H.; Cui, X.Y.; Gao, C.Y. Hypoxic preconditioning: A novel intrinsic cytoprotective strategy. Mol. Neurobiol. 2005, 31, 255–271. [Google Scholar] [CrossRef]
- Vangeison, G.; Carr, D.; Federoff, H.J.; Rempe, D.A. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J. Neurosci. 2008, 28, 1988–1993. [Google Scholar] [CrossRef]
- Wang, F.Z.; Ding, A.S. Protective effect of calcitonin gene-related peptide on synaptic function in hippocampal slice during hypoxia. Sheng Li Xue Bao 1994, 46, 529–538. (In Chinese) [Google Scholar]
- Jones, S.M.; Novak, A.E.; Elliott, J.P. The role of HIF in cobalt-induced ischemic tolerance. Neuroscience 2013, 252C, 420–430. [Google Scholar] [CrossRef]
- Jones, N.M.; Kardashyan, L.; Callaway, J.K.; Lee, E.M.; Beart, P.M. Long-term functional and protective actions of preconditioning with hypoxia, cobalt chloride, and desferrioxamine against hypoxic-ischemic injury in neonatal rats. Pediatr. Res. 2008, 63, 620–624. [Google Scholar] [CrossRef]
- Ruscher, K.; Isaev, N.; Trendelenburg, G.; Weih, M.; Iurato, L.; Meisel, A.; Dirnagl, U. Induction of hypoxia inducible factor 1 by oxygen glucose deprivation is attenuated by hypoxic preconditioning in rat cultured neurons. Neurosci. Lett. 1998, 254, 117–120. [Google Scholar] [CrossRef]
- Camenisch, G.; Stroka, D.M.; Gassmann, M.; Wenger, R.H. Attenuation of HIF-1 DNA-binding activity limits hypoxia-inducible endothelin-1 expression. Pflüg. Arch. 2001, 443, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Sakanaka, M.; Wen, T.C.; Matsuda, S.; Masuda, S.; Morishita, E.; Nagao, M.; Sasaki, R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 1998, 95, 4635–4640. [Google Scholar] [CrossRef]
- Marti, H.J.; Bernaudin, M.; Bellail, A.; Schoch, H.; Euler, M.; Petit, E.; Risau, W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am. J. Pathol. 2000, 156, 965–976. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Sejas, D.P.; Bagby, G.C.; Pang, Q. Hypoxia-induced nucleophosmin protects cell death through inhibition of p53. J. Biol. Chem. 2004, 279, 41275–41279. [Google Scholar]
- Jones, N.M.; Bergeron, M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J. Cereb. Blood Flow Metab. 2001, 21, 1105–1114. [Google Scholar] [CrossRef]
- Fairchild, M.D.; Parsons, J.E.; Wasterlain, C.G.; Rinaldi, P.C.; Wallis, R.A. A hypoxic injury potential in the hippocampal slice. Brain Res. 1988, 453, 357–361. [Google Scholar] [CrossRef]
- Burmester, T.; Gerlach, F.; Hankeln, T. Regulation and role of neuroglobin and cytoglobin under hypoxia. Adv. Exp. Med. Biol. 2007, 618, 169–180. [Google Scholar] [CrossRef]
- Rey, S.; Semenza, G.L. Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef]
- Benita, Y.; Kikuchi, H.; Smith, A.D.; Zhang, M.Q.; Chung, D.C.; Xavier, R.J. An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009, 37, 4587–4602. [Google Scholar]
- Schmedtje, J.F., Jr.; Ji, Y.S.; Liu, W.L.; DuBois, R.N.; Runge, M.S. Hypoxia induces cyclooxygenase-2 via the NF-κB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 1997, 272, 601–608. [Google Scholar]
- Dong, Z.; Venkatachalam, M.A.; Wang, J.; Patel, Y.; Saikumar, P.; Semenza, G.L.; Force, T.; Nishiyama, J. Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia. HIF-1-independent mechanisms. J. Biol. Chem. 2001, 276, 18702–18709. [Google Scholar] [CrossRef]
- Hofer, T.; Wenger, R.H.; Kramer, M.F.; Ferreira, G.C.; Gassmann, M. Hypoxic up-regulation of erythroid 5-aminolevulinate synthase. Blood 2003, 101, 348–350. [Google Scholar] [CrossRef]
- Dong, Z.; Wang, J.Z.; Yu, F.; Venkatachalam, M.A. Apoptosis-resistance of hypoxic cells: Multiple factors involved and a role for IAP-2. Am. J. Pathol. 2003, 163, 663–671. [Google Scholar] [CrossRef]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Todorov, V.; Gess, B.; Godecke, A.; Wagner, C.; Schrader, J.; Kurtz, A. Endogenous nitric oxide attenuates erythropoietin gene expression in vivo. Pflüg. Arch. 2000, 439, 445–448. [Google Scholar] [CrossRef]
- Chung, J.W.; Shin, J.E.; Han, K.W.; Ahn, J.H.; Kim, Y.J.; Park, J.W.; So, H.S. Up-regulation of hypoxia-inducible factor-1 alpha by cobalt chloride prevents hearing loss in noise-exposed mice. Environ. Toxicol. Pharmacol. 2011, 31, 153–159. [Google Scholar] [CrossRef]
- Bergeron, M.; Gidday, J.M.; Yu, A.Y.; Semenza, G.L.; Ferriero, D.M.; Sharp, F.R. Role of hypoxia-inducible factor-1 in hypoxia-induced ischemic tolerance in neonatal rat brain. Ann. Neurol. 2000, 48, 285–296. [Google Scholar]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y.-B.; Wang, X.; Meister, E.A.; Gong, K.-R.; Yan, S.-C.; Lu, G.-W.; Ji, X.-M.; Shao, G. The Effects of CoCl2 on HIF-1α Protein under Experimental Conditions of Autoprogressive Hypoxia Using Mouse Models. Int. J. Mol. Sci. 2014, 15, 10999-11012. https://doi.org/10.3390/ijms150610999
Zhang Y-B, Wang X, Meister EA, Gong K-R, Yan S-C, Lu G-W, Ji X-M, Shao G. The Effects of CoCl2 on HIF-1α Protein under Experimental Conditions of Autoprogressive Hypoxia Using Mouse Models. International Journal of Molecular Sciences. 2014; 15(6):10999-11012. https://doi.org/10.3390/ijms150610999
Chicago/Turabian StyleZhang, Yan-Bo, Xiulian Wang, Edward A. Meister, Ke-Rui Gong, Shao-Chun Yan, Guo-Wei Lu, Xun-Ming Ji, and Guo Shao. 2014. "The Effects of CoCl2 on HIF-1α Protein under Experimental Conditions of Autoprogressive Hypoxia Using Mouse Models" International Journal of Molecular Sciences 15, no. 6: 10999-11012. https://doi.org/10.3390/ijms150610999