TRAF6 Inhibition Rescues Dexamethasone-Induced Muscle Atrophy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
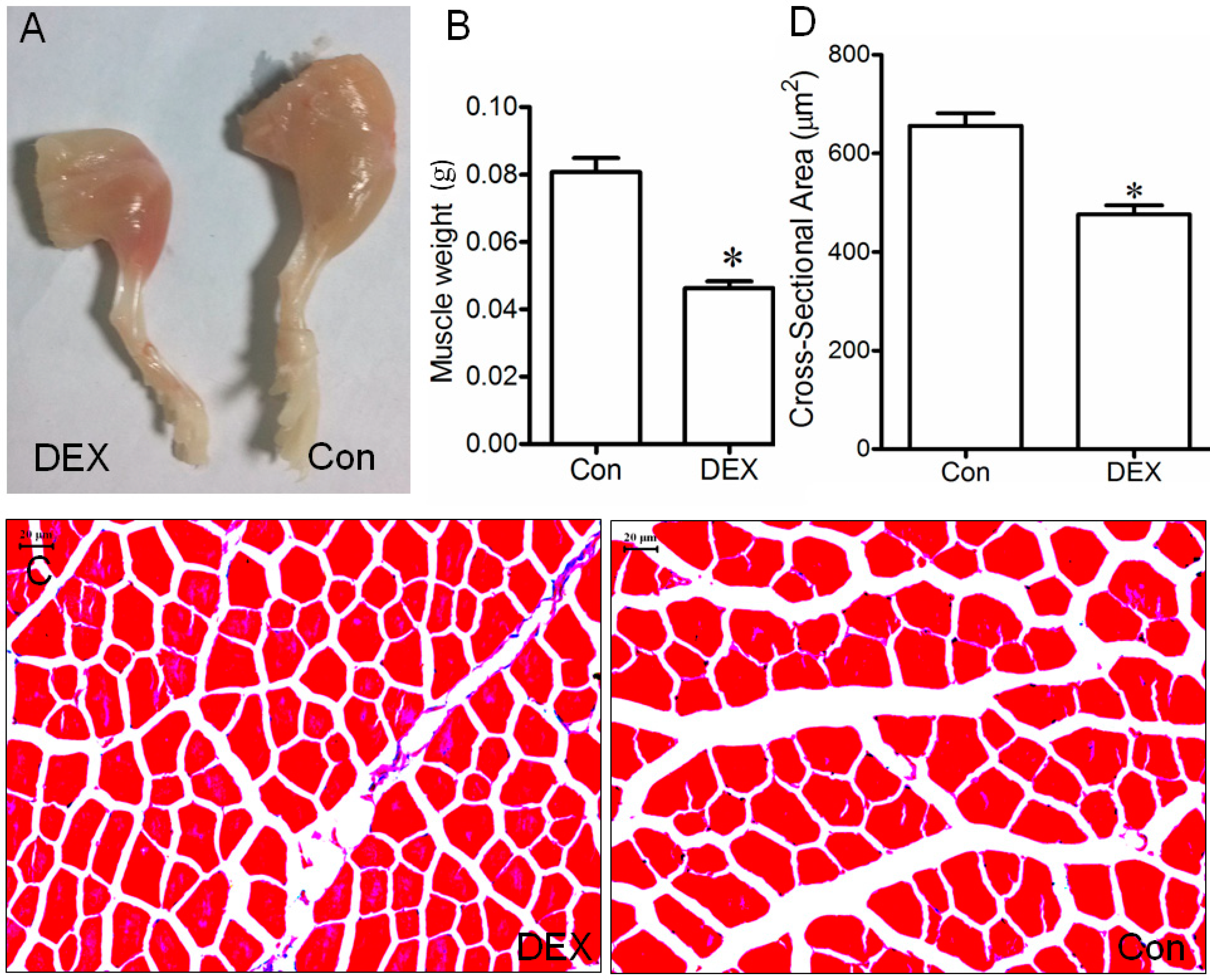
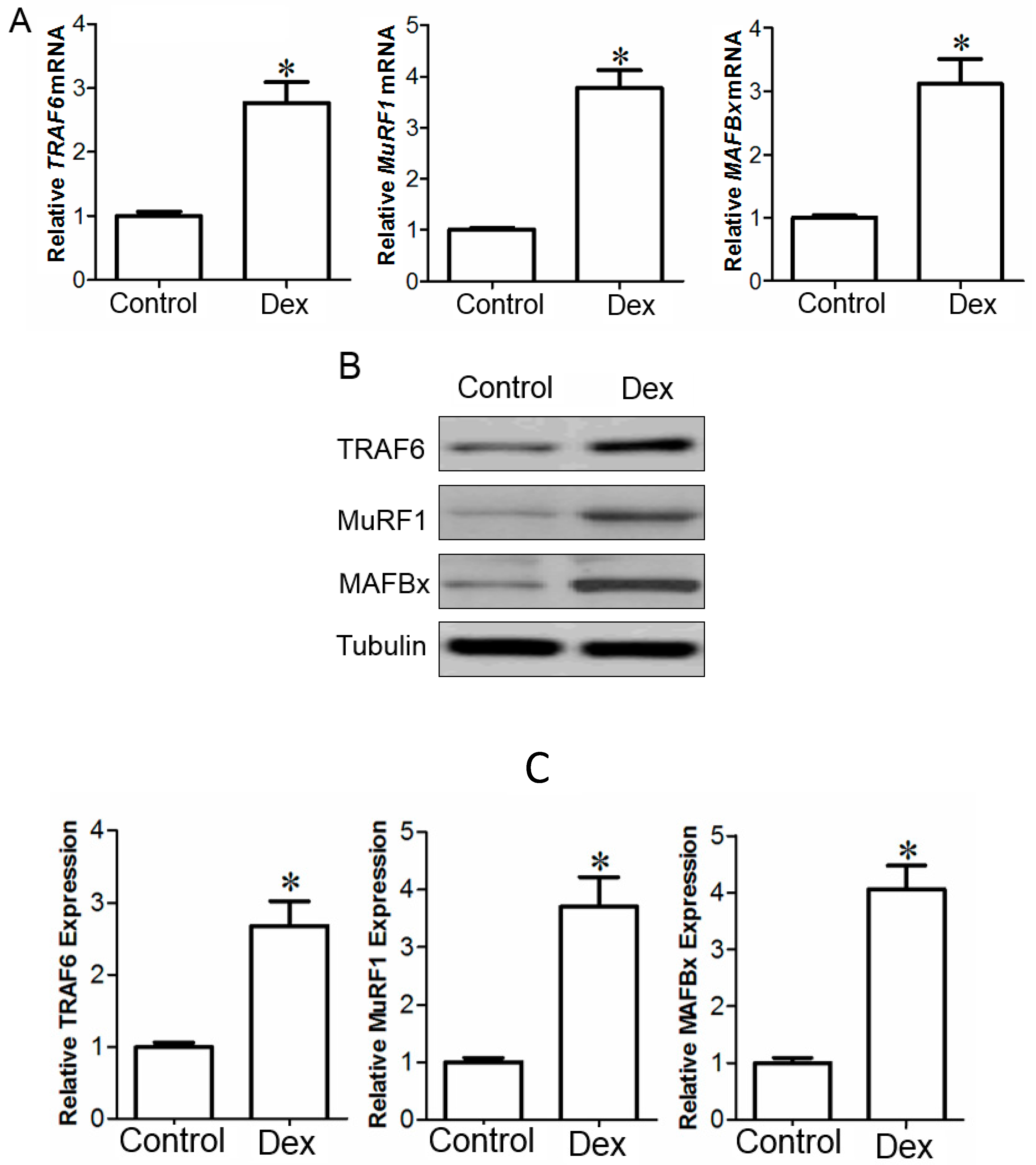
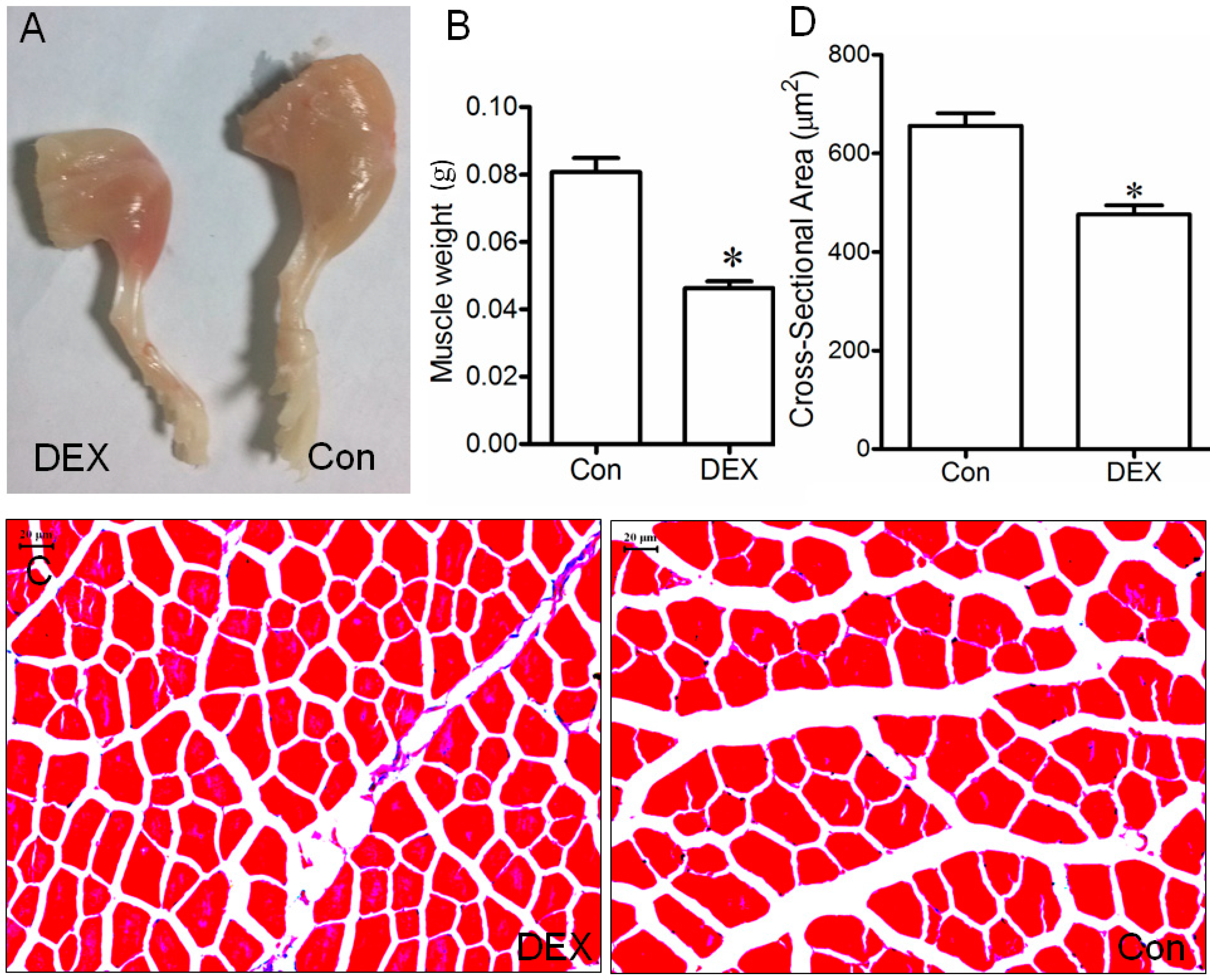
2.1. Up-Regulation of Tumor Necrosis Factor Receptor-Associated Factor 6 (TRAF6), Muscle Atrophy F-Box (MAFBx), and Muscle Ring Finger 1 (MuRF1) in Muscle and Myotube Treated with Dex

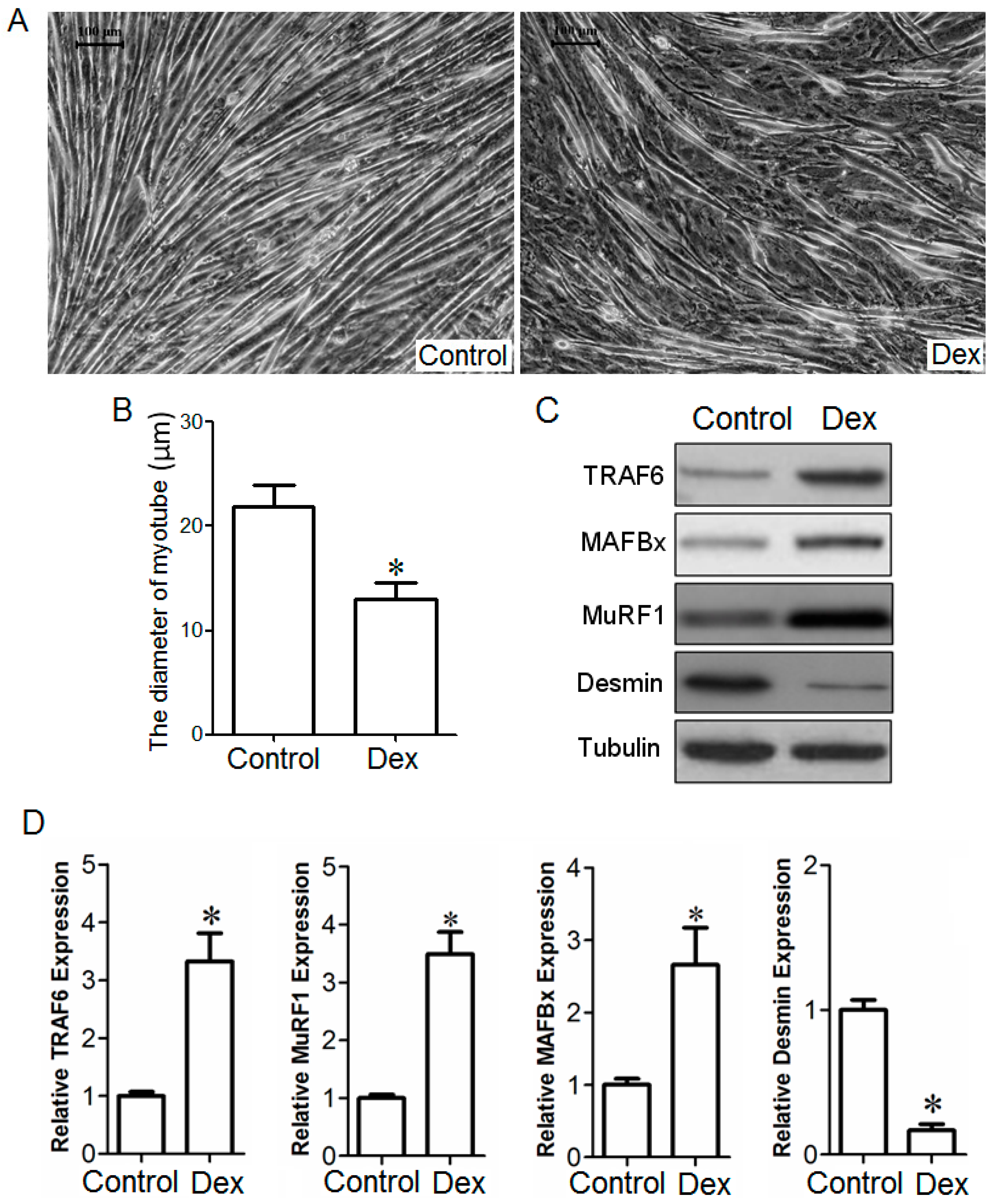
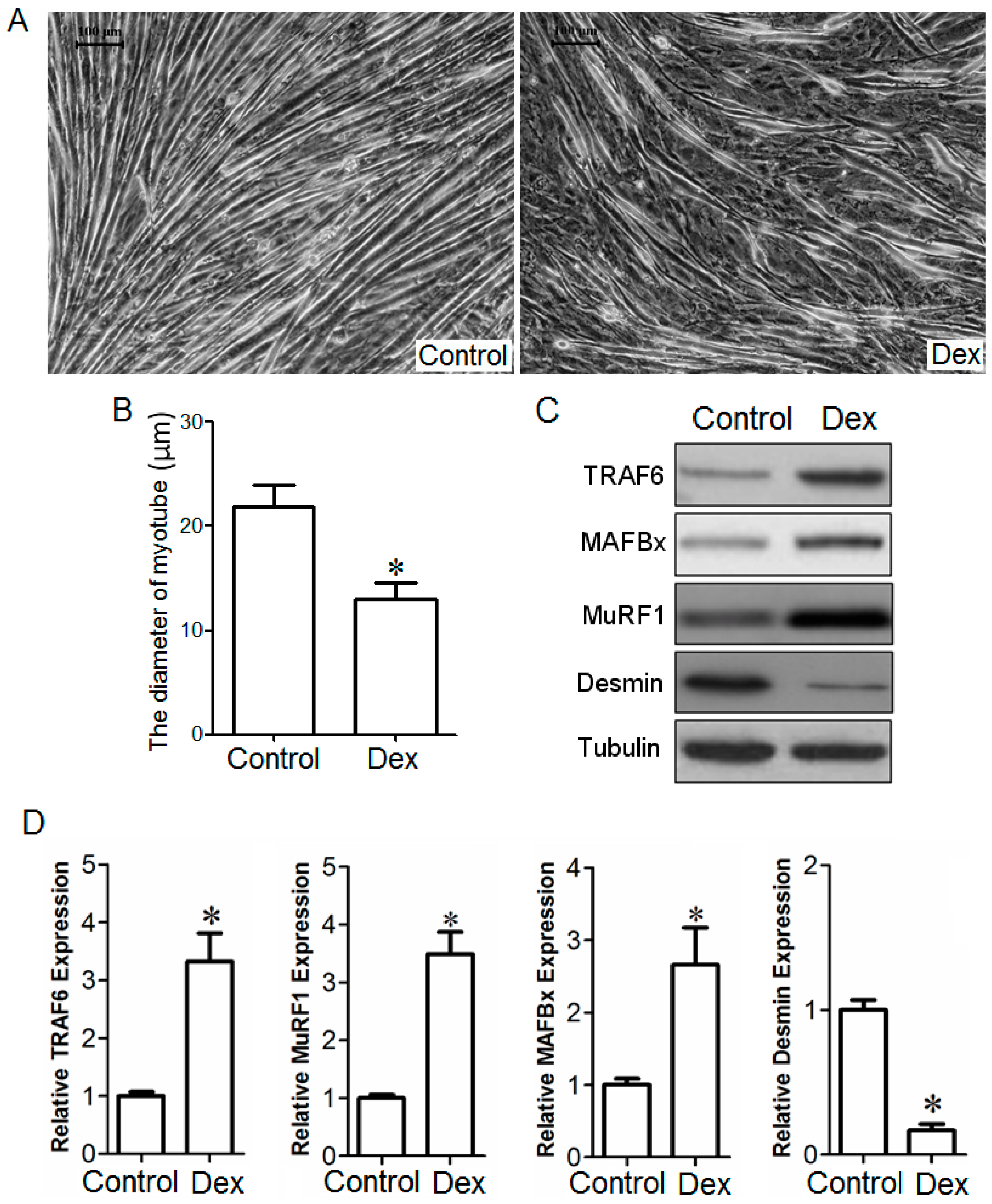
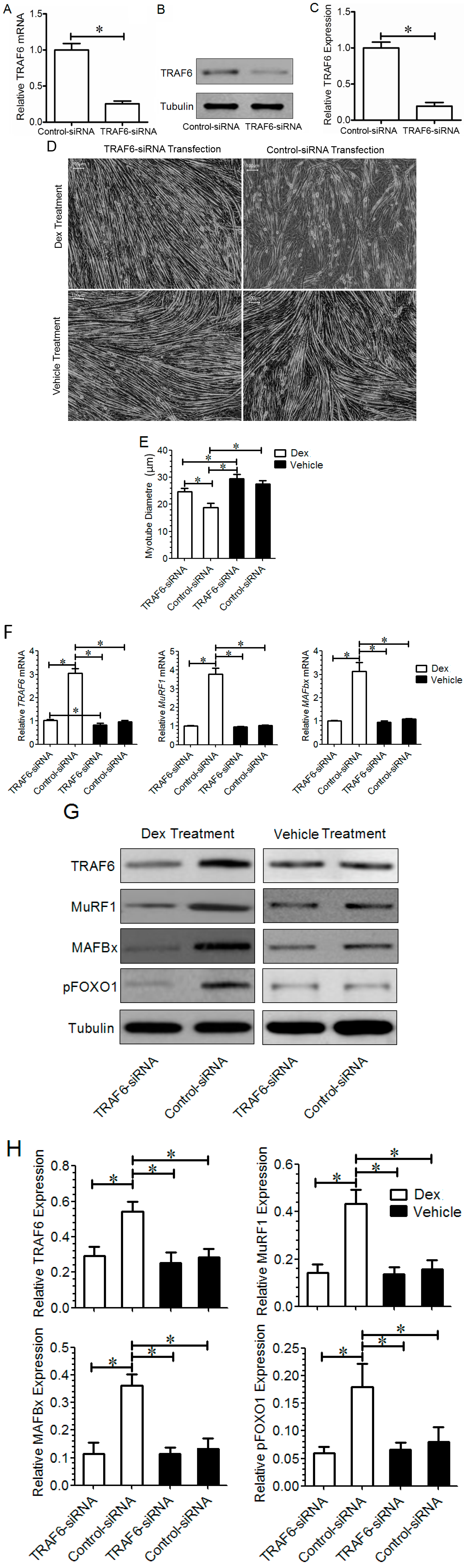
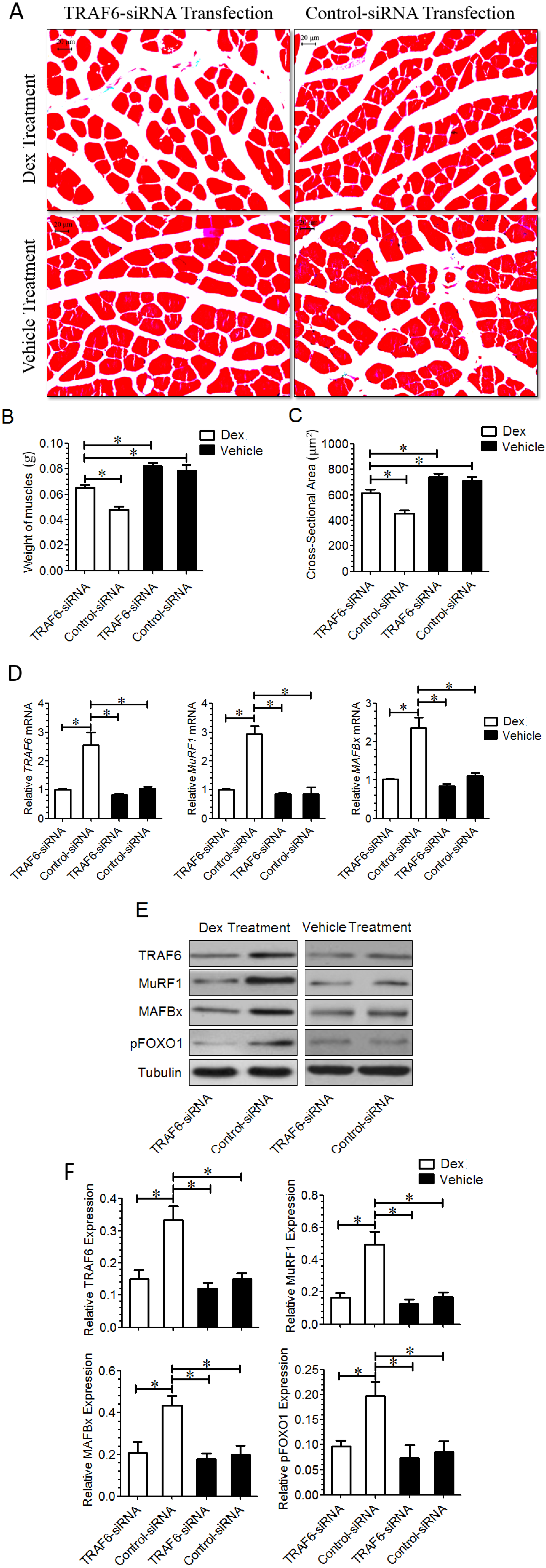
2.2. Effect of TRAF6 Knockdown on Dex-Induced Myotube Atrophy in Vitro
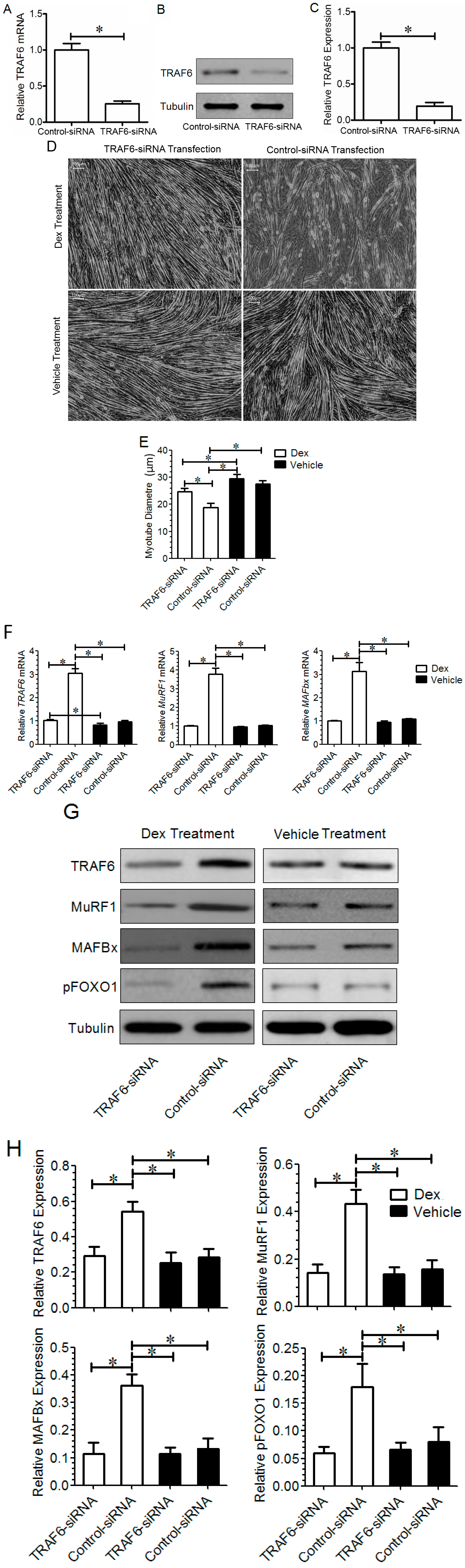
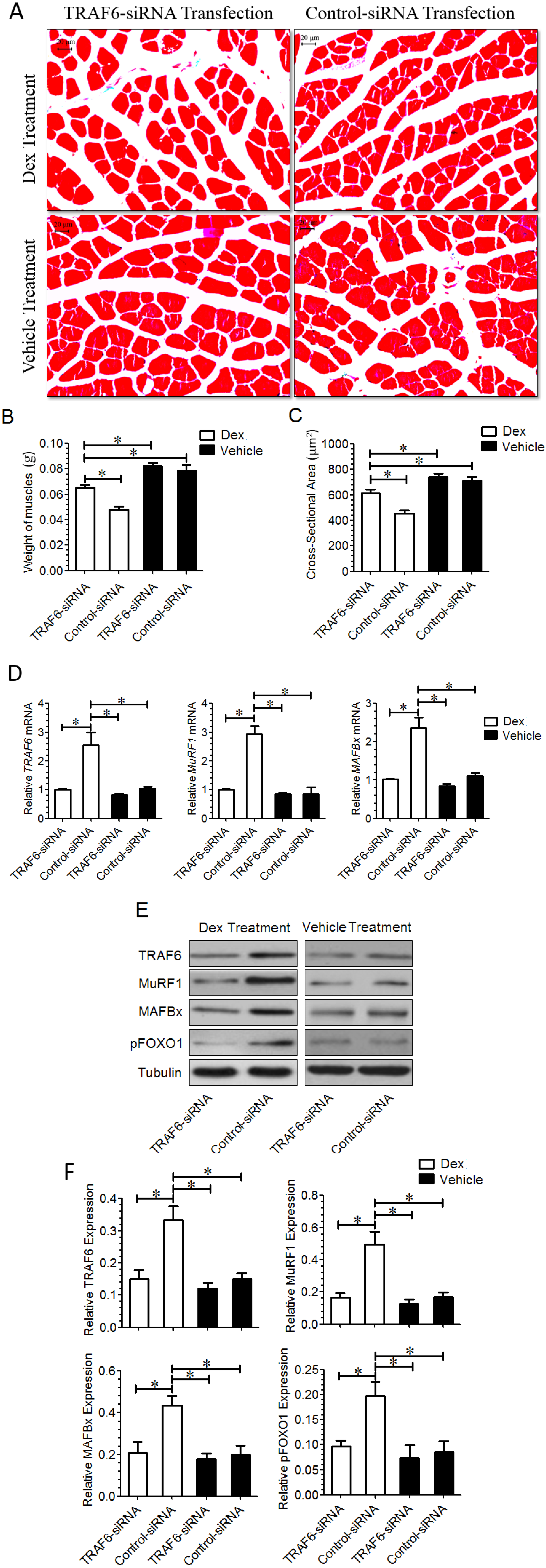
2.3. Effect of TRAF6 Knockdown on Dex-Induced Muscle Atrophy in Vivo
3. Discussion
4. Experimental Section
4.1. Animal Care and Treatments
4.2. Cell Culture and Transfection
4.3. Real-Time Quantitative RT-PCR (qPCR)
4.4. Western Blot Analysis
4.5. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Jackman, R.W.; Kandarian, S.C. The molecular basis of skeletal muscle atrophy. Am. J. Physiol. Cell Physiol. 2004, 287, C834–C843. [Google Scholar] [CrossRef]
- Piccirillo, R.; Demontis, F.; Perrimon, N.; Goldberg, A.L. Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev. Dyn. 2014, 243, 201–215. [Google Scholar] [CrossRef]
- Piccirillo, R.; Goldberg, A.L. The p97/VCP ATPase is critical in muscle atrophy and the accelerated degradation of muscle proteins. EMBO J. 2012, 31, 3334–3350. [Google Scholar] [CrossRef]
- Jones, A.; Hwang, D.J.; Narayanan, R.; Miller, D.D.; Dalton, J.T. Effects of a novel selective androgen receptor modulator on dexamethasone-induced and hypogonadism-induced muscle atrophy. Endocrinology 2010, 151, 3706–3719. [Google Scholar] [CrossRef]
- Ma, K.; Mallidis, C.; Bhasin, S.; Mahabadi, V.; Artaza, J.; Gonzalez-Cadavid, N.; Arias, J.; Salehian, B. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am. J. Physiol. Endocrinol. Metabol. 2003, 285, E363–E371. [Google Scholar]
- Kuo, T.; Harris, C.A.; Wang, J.C. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol. Cell. Endocrinol. 2013, 380, 79–88. [Google Scholar] [CrossRef]
- Menconi, M.; Gonnella, P.; Petkova, V.; Lecker, S.; Hasselgren, P.O. Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J. Cell. Biochem. 2008, 105, 353–364. [Google Scholar] [CrossRef]
- Zhao, W.; Pan, J.; Zhao, Z.; Wu, Y.; Bauman, W.A.; Cardozo, C.P. Testosterone protects against dexamethasone-induced muscle atrophy, protein degradation and MAFbx upregulation. J. Steroid Biochem. Mol. Biol. 2008, 110, 125–129. [Google Scholar] [CrossRef]
- Aversa, Z.; Alamdari, N.; Castillero, E.; Muscaritoli, M.; Rossi Fanelli, F.; Hasselgren, P.O. β-Hydroxy-β-methylbutyrate (HMB) prevents dexamethasone-induced myotube atrophy. Biochem. Biophy. Res. Commun. 2012, 423, 739–743. [Google Scholar] [CrossRef]
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle sparing in muscle RING finger 1 null mice: Response to synthetic glucocorticoids. J. Physiol. 2011, 589, 4759–4776. [Google Scholar]
- Schakman, O.; Gilson, H.; Kalista, S.; Thissen, J.P. Mechanisms of muscle atrophy induced by glucocorticoids. Horm. Res. 2009, 72, 36–41. [Google Scholar]
- Wu, H.; Arron, J.R. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. Cell. Dev. Biol. 2003, 25, 1096–1105. [Google Scholar]
- Zhang, X.; Zhang, J.; Zhang, L.; van Dam, H.; ten Dijke, P. UBE2O negatively regulates TRAF6-mediated NF-κB activation by inhibiting TRAF6 polyubiquitination. Cell Res. 2013, 23, 366–377. [Google Scholar] [CrossRef]
- Paul, P.K.; Gupta, S.K.; Bhatnagar, S.; Panguluri, S.K.; Darnay, B.G.; Choi, Y.; Kumar, A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol. 2010, 191, 1395–1411. [Google Scholar] [CrossRef]
- Paul, P.K.; Bhatnagar, S.; Mishra, V.; Srivastava, S.; Darnay, B.G.; Choi, Y.; Kumar, A. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol.Cell. Biol. 2012, 32, 1248–1259. [Google Scholar] [CrossRef]
- Nakamura, K.; Kimple, A.J.; Siderovski, D.P.; Johnson, G.L. PB1 domain interaction of p62/sequestosome 1 and MEKK3 regulates NF-κB activation. J. Biol. Chem. 2010, 285, 2077–2089. [Google Scholar]
- Lamothe, B.; Campos, A.D.; Webster, W.K.; Gopinathan, A.; Hur, L.; Darnay, B.G. The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL. J. Biol. Chem. 2008, 283, 24871–24880. [Google Scholar]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar]
- Paul, P.K.; Kumar, A. TRAF6 coordinates the activation of autophagy and ubiquitin-proteasome systems in atrophying skeletal muscle. Autophagy 2011, 7, 555–556. [Google Scholar] [CrossRef]
- Tiao, G.; Fagan, J.; Roegner, V.; Lieberman, M.; Wang, J.J.; Fischer, J.E.; Hasselgren, P.O. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J. Clin. Investig. 1996, 97, 339–348. [Google Scholar] [CrossRef]
- Aharinejad, S.; Paulus, P.; Sioud, M.; Hofmann, M.; Zins, K.; Schafer, R.; Stanley, E.R.; Abraham, D. Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice. Cancer Res. 2004, 64, 5378–5384. [Google Scholar] [CrossRef]
- Sun, H.; Qiu, J.; Chen, Y.; Yu, M.; Ding, F.; Gu, X. Proteomic and bioinformatic analysis of differentially expressed proteins in denervated skeletal muscle. Int. J. Mol. Med. 2014, 33, 1586–1596. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, T.; Ding, F.; Hu, N.; Gu, X. Proteomic studies of rat tibialis anterior muscle during postnatal growth and development. Mol. Cell. Biochem. 2009, 332, 161–171. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sun, H.; Gong, Y.; Qiu, J.; Chen, Y.; Ding, F.; Zhao, Q. TRAF6 Inhibition Rescues Dexamethasone-Induced Muscle Atrophy. Int. J. Mol. Sci. 2014, 15, 11126-11141. https://doi.org/10.3390/ijms150611126
Sun H, Gong Y, Qiu J, Chen Y, Ding F, Zhao Q. TRAF6 Inhibition Rescues Dexamethasone-Induced Muscle Atrophy. International Journal of Molecular Sciences. 2014; 15(6):11126-11141. https://doi.org/10.3390/ijms150611126
Chicago/Turabian StyleSun, Hualin, Yanpei Gong, Jiaying Qiu, Yanfei Chen, Fei Ding, and Qing Zhao. 2014. "TRAF6 Inhibition Rescues Dexamethasone-Induced Muscle Atrophy" International Journal of Molecular Sciences 15, no. 6: 11126-11141. https://doi.org/10.3390/ijms150611126