A Simple Three-Step Method for Design and Affinity Testing of New Antisense Peptides: An Example of Erythropoietin

 ,
,
Abstract
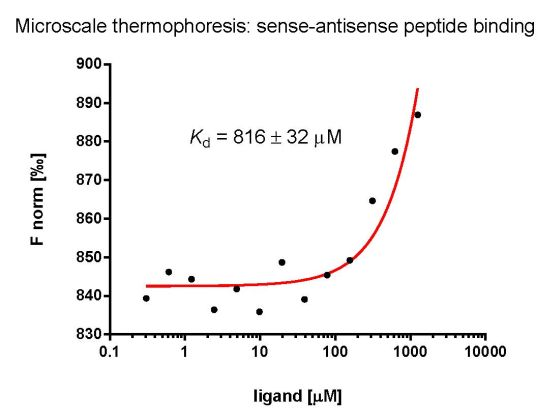
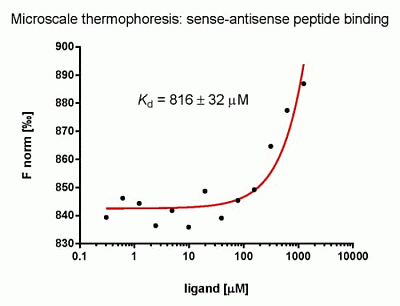
:
1. Introduction
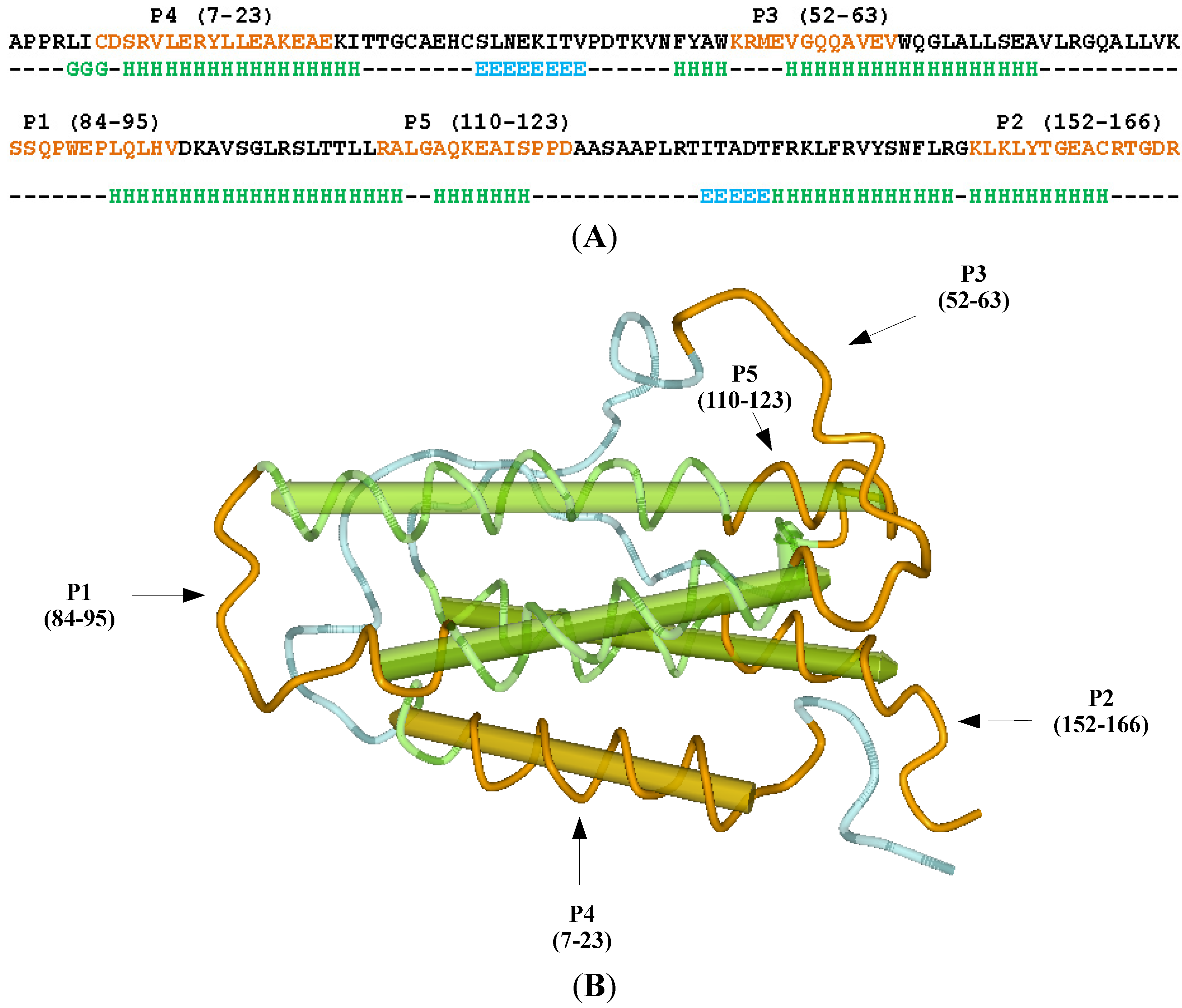
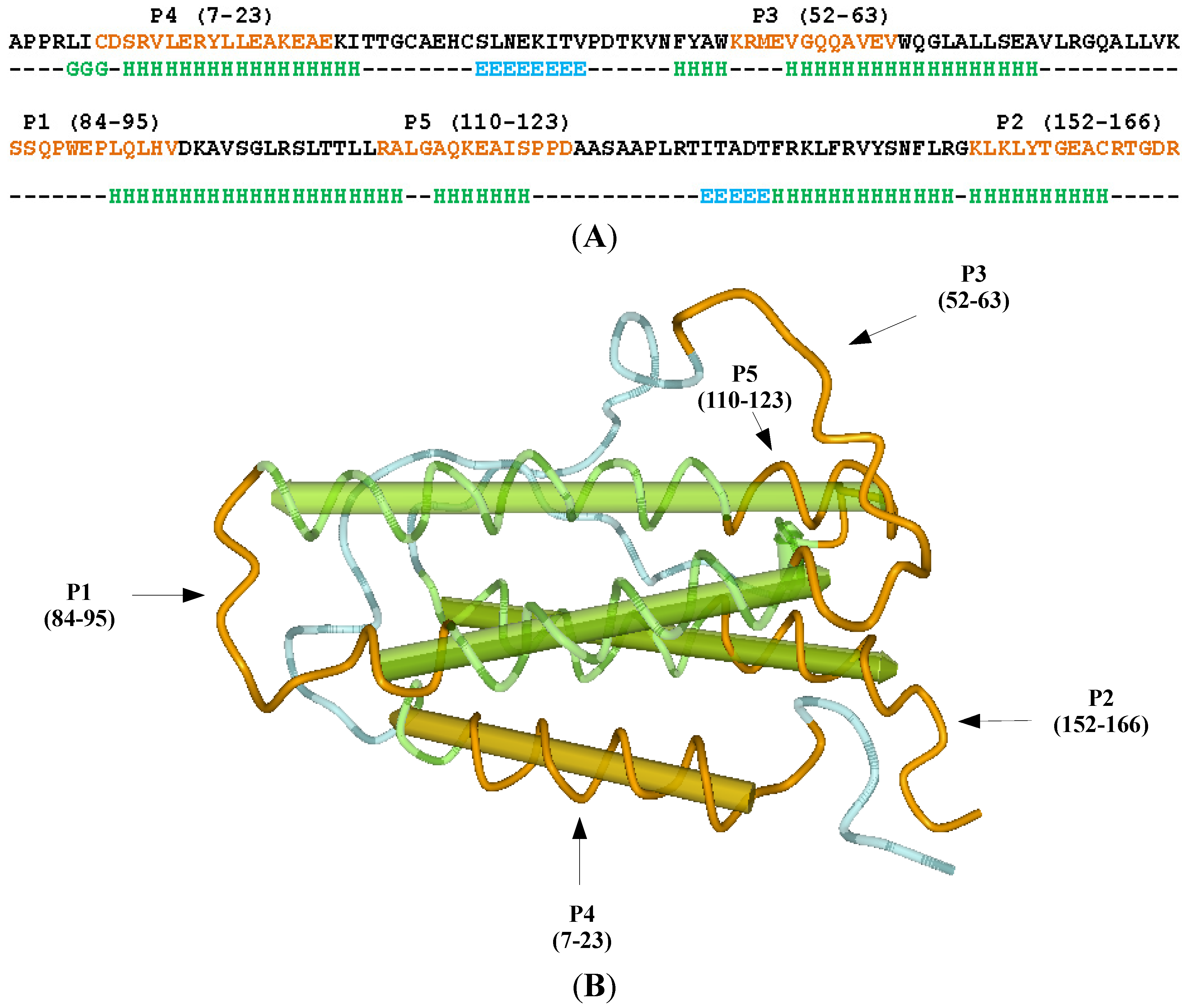
2. Results and Discussion
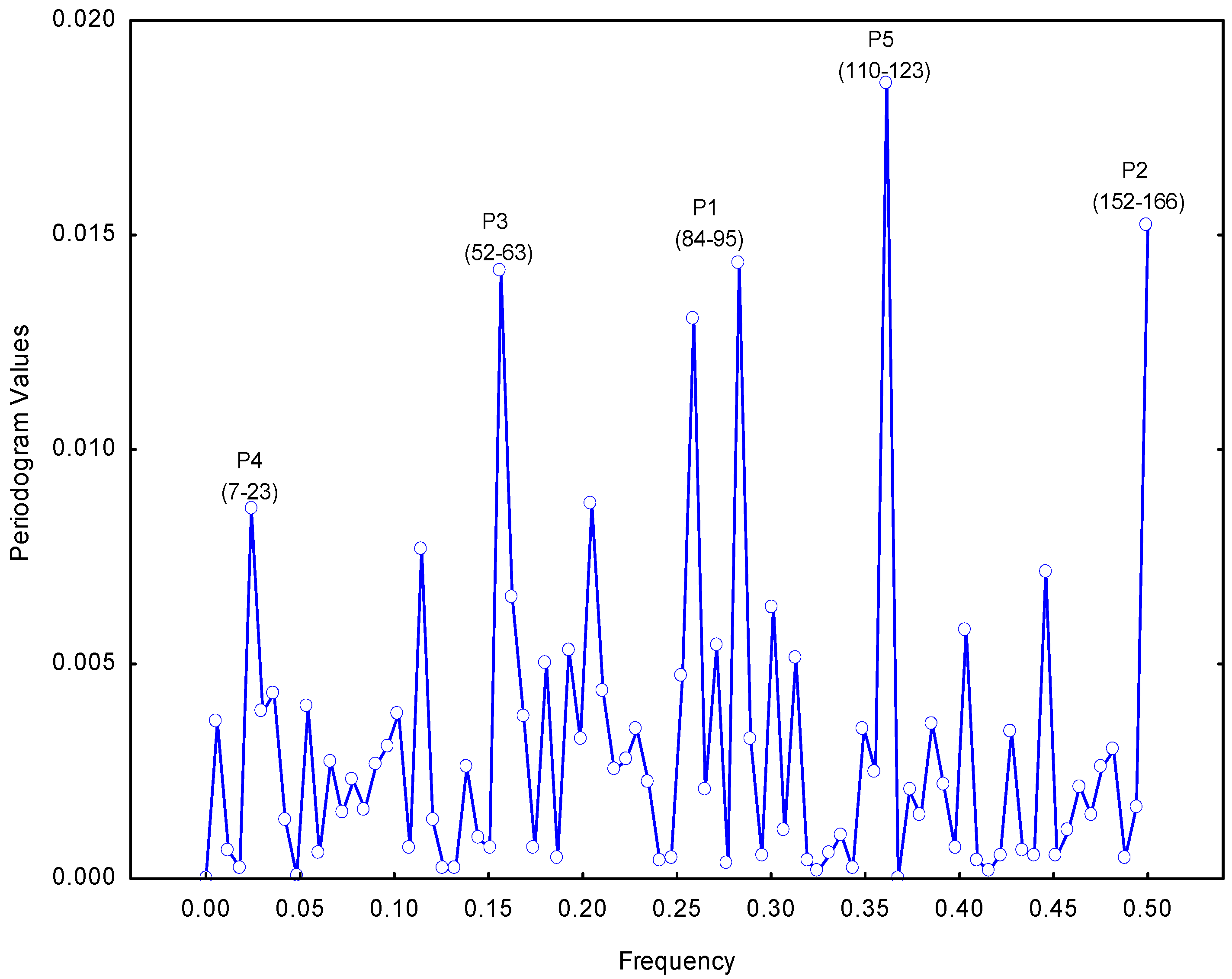
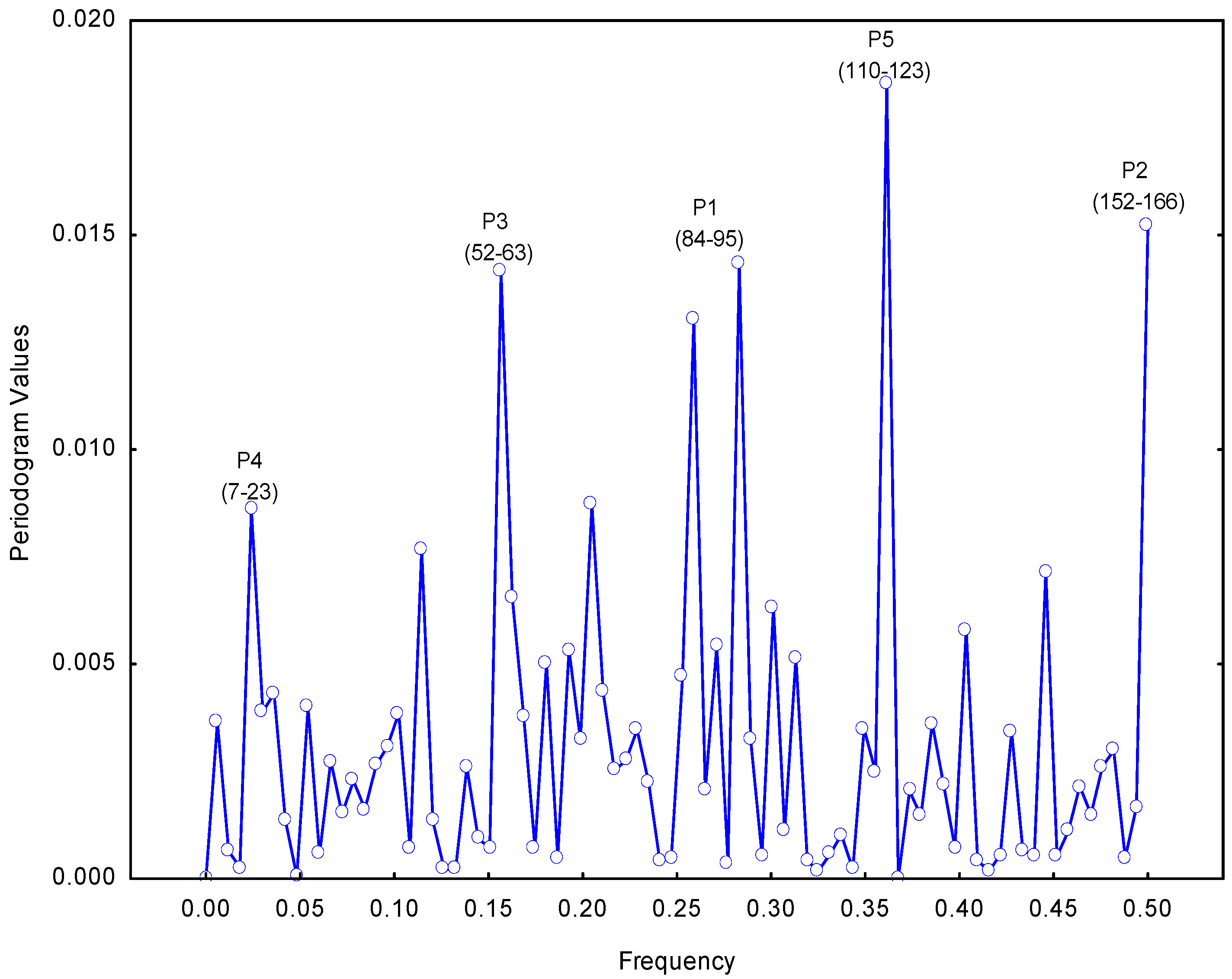
2.1. Selection of the Molecular Target (Epitope): Step 1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid | Abbreviation | Hopp and Woods Hydrophilicity | EIIP (Ry) |
|---|---|---|---|
| Arginine | R | 3.0 | 0.0959 |
| Lysine | K | 3.0 | 0.0371 |
| Aspartic acid | D | 3.0 | 0.1263 |
| Glutamic acid | E | 3.0 | 0.0058 |
| Serine | S | 0.3 | 0.0829 |
| Asparagine | N | 0.2 | 0.0036 |
| Glutamine | Q | 0.2 | 0.0761 |
| Proline | P | 0.0 | 0.0198 |
| Glycine | G | 0.0 | 0.0050 |
| Threonine | T | −0.4 | 0.0941 |
| Histidine | H | −0.5 | 0.0242 |
| Alanine | A | −0.5 | 0.0373 |
| Cysteine | C | −1.0 | 0.0829 |
| Methionine | M | −1.3 | 0.0823 |
| Valine | V | −1.5 | 0.0057 |
| Leucine | L | −1.8 | 0.0000 |
| Isoleucine | I | −1.8 | 0.0000 |
| Tyrosine | Y | −2.3 | 0.0516 |
| Phenylalanine | F | −2.5 | 0.0946 |
| Tryptophan | W | −3.4 | 0.0548 |
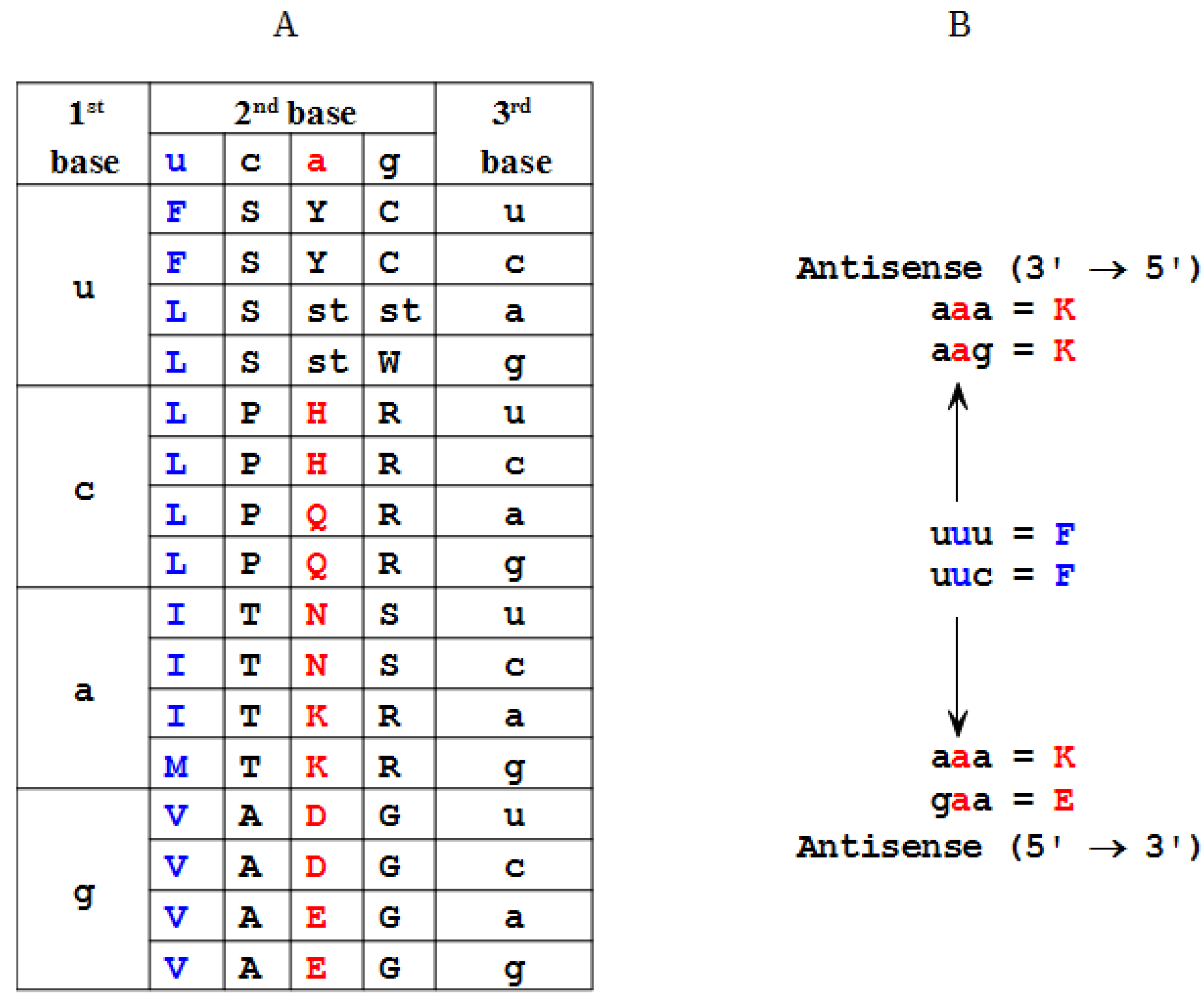
2.2. Modeling of an Ansisense Peptide (Paratope): Step 2
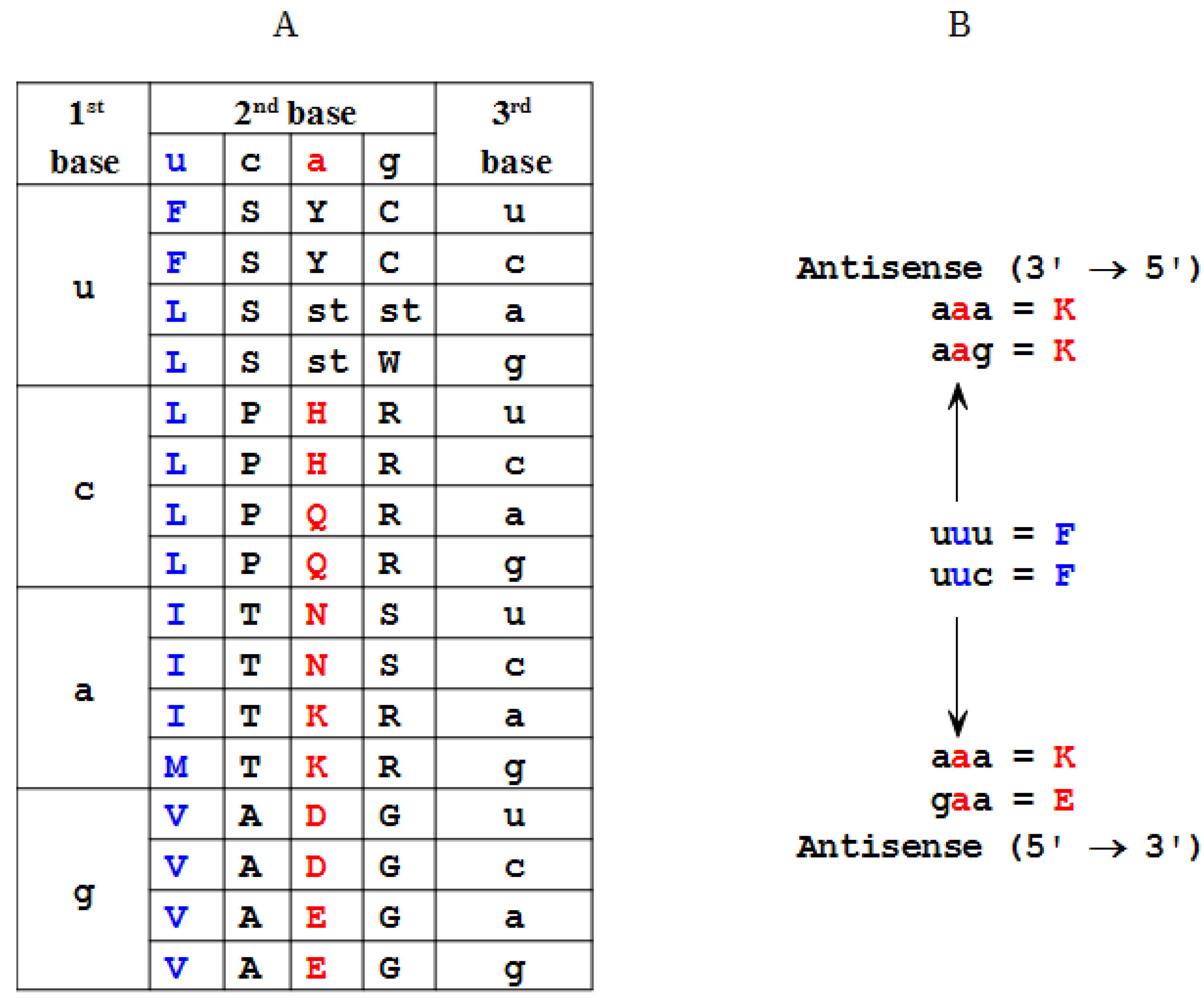
| Amino Acid | Antisense 3'→5' | Antisense 5'→3' |
|---|---|---|
| F | K | K, E (η) |
| L | D, E, N (α) | E, Q, K (θ) |
| I | Y | N, D, Y (ι) |
| M | Y | H |
| V | H, Q (β) | H, D, N, Y (κ) |
| S | S, R (γ) | G, R, T, A (λ) |
| P | G | G, W, R (μ) |
| T | W, C (δ) | G, S, C, R (ν) |
| A | R | R, G, S, C (ξ) |
| Y | M, I (ε) | I, V (ο) |
| H | V | V, M (π) |
| Q | V | L |
| N | L | I, V (ρ) |
| K | F | F, L (ς) |
| D | L | I, V (σ) |
| E | L | L, F (τ) |
| C | T | T, A (υ) |
| W | T | P |
| R | A, S (ζ) | A, S, P, T (φ) |
| G | P | P, S, T, A (χ) |
| Antisense Paratope | EPO-P2 Epitope KLFLYTGEACRTGDR | Number of Paratopes | BLAST Detected Antibodies |
|---|---|---|---|
| 1 | FαFαε | 18 | 6 |
| 2 | αFαεδ | 36 | 20 |
| 3 | FαεδP | 12 | 0 |
| 4 | αεδPL | 12 | 0 |
| 5 | εδPLR | 4 | 0 |
| 6 | δPLRT | 2 | 0 |
| 7 | PLRTζ | 4 | 2 |
| 8 | LRTζδ | 4 | 0 |
| 9 | RTζδP | 4 | 4 |
| 10 | TζδPL | 4 | 6 |
| 11 | ζδPLζ | 8 | 12 |
| Antisense Paratope 2 | Antisense Paratope 7 | Antisense Paratope 11 | BLAST Detected Antibodies |
|---|---|---|---|
| DFDIW | PLRTA | WPLS | 17 |
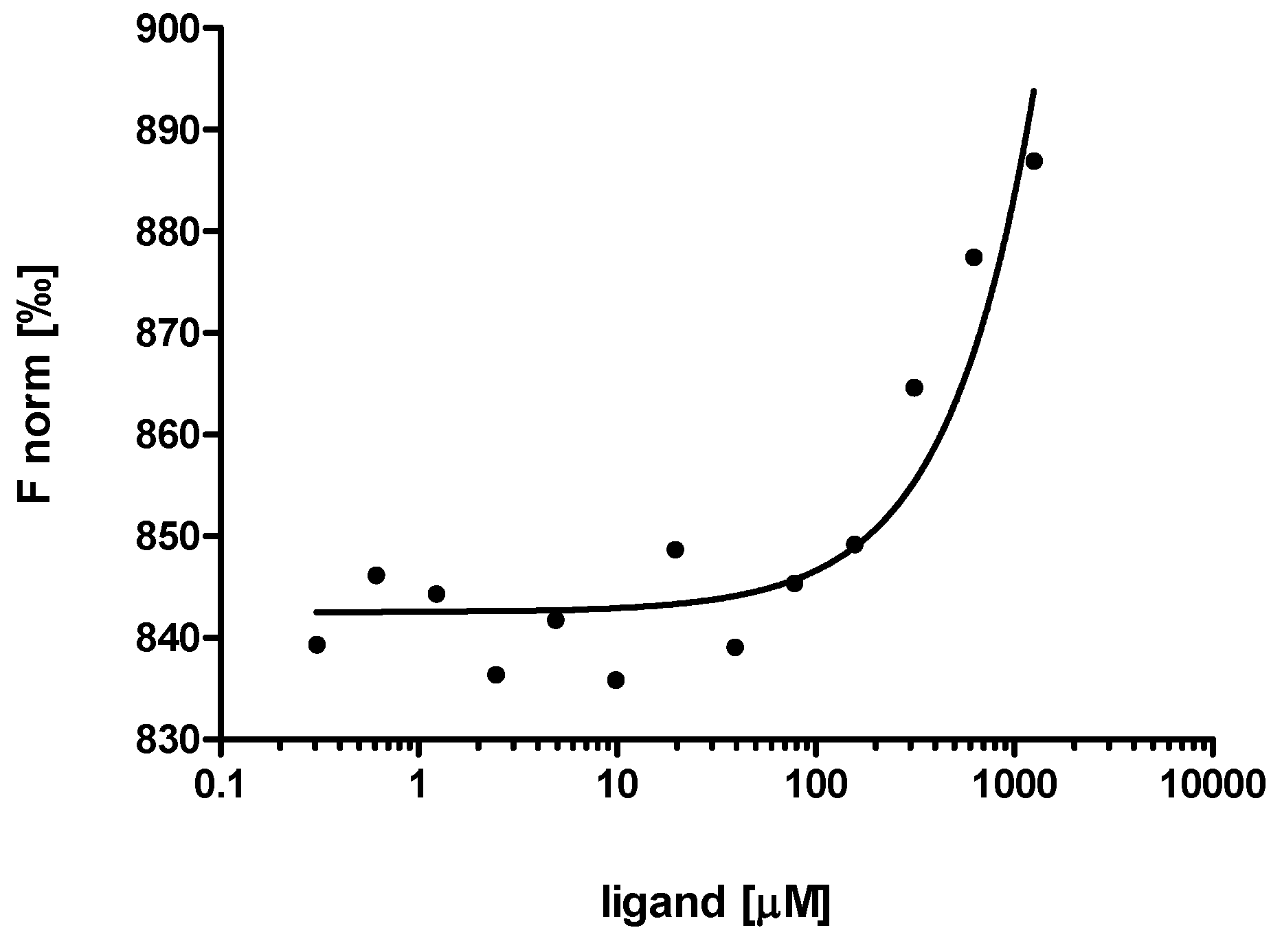
2.3. Evaluation of Peptide Binding: Step 3
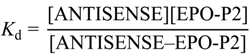
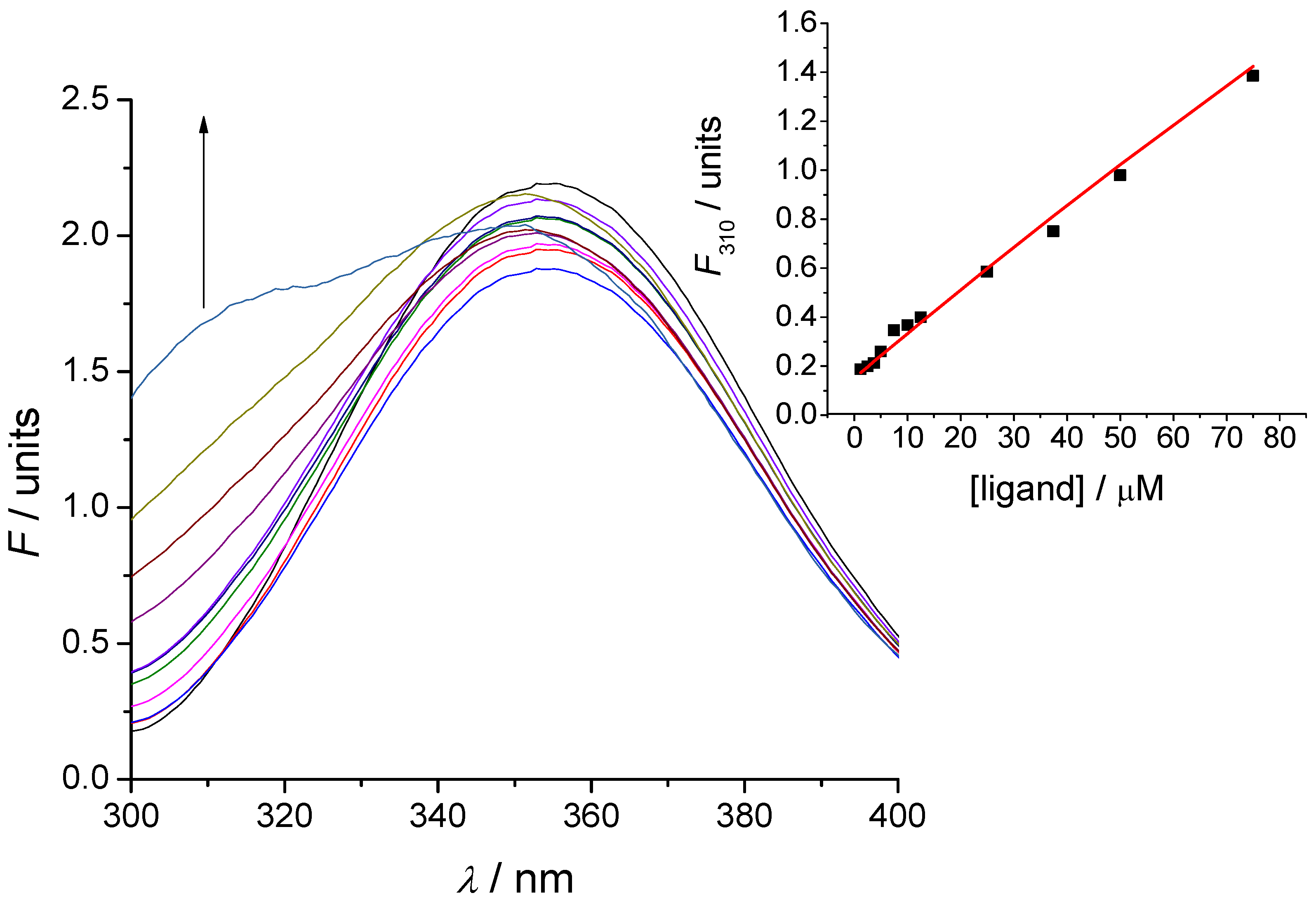
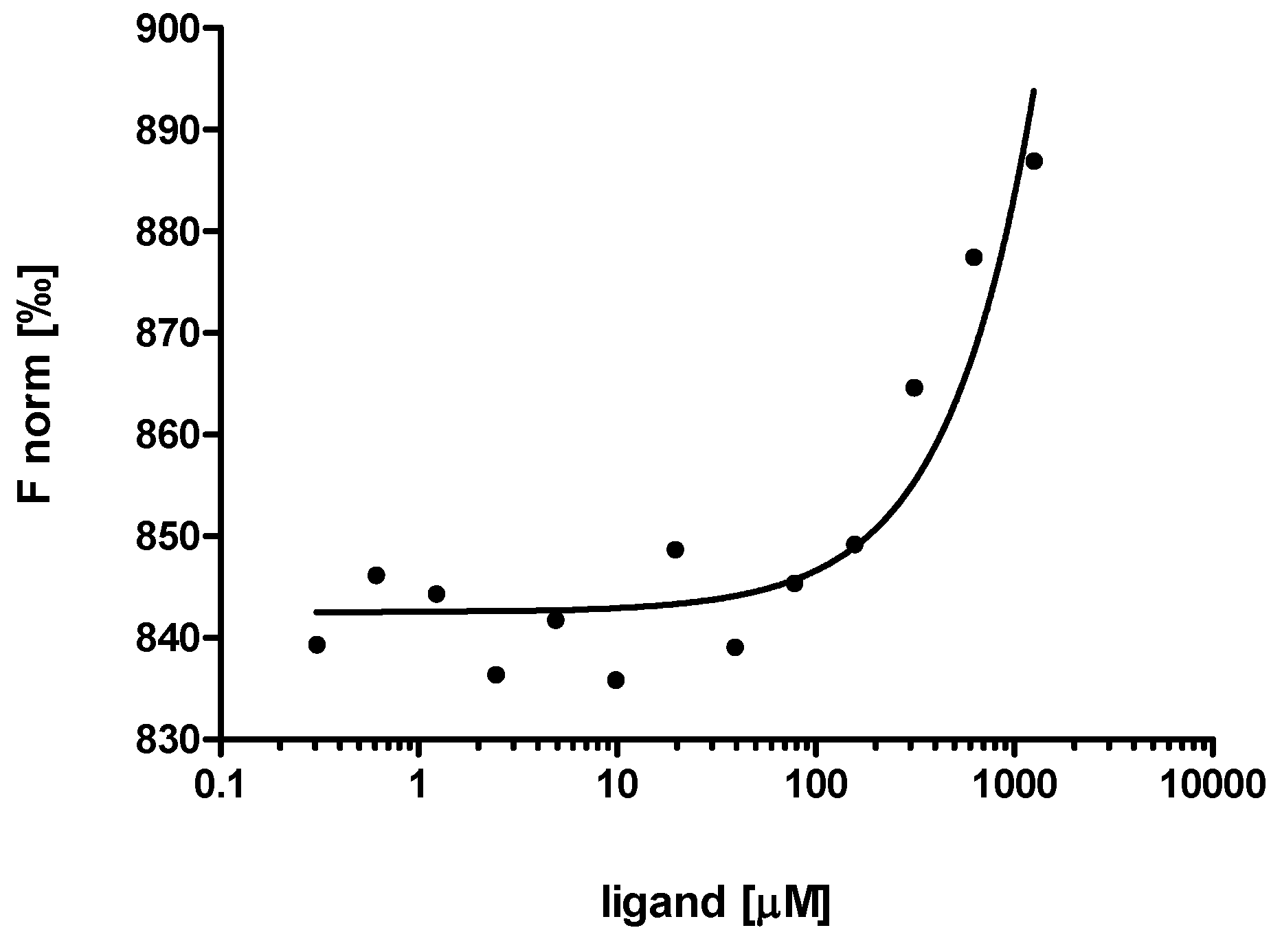
3. Experimental Section
3.1. Erythropoietin (EPO) Molecule Structure
3.2. Resonant Recognition Method (RRM)
3.3. Peptides
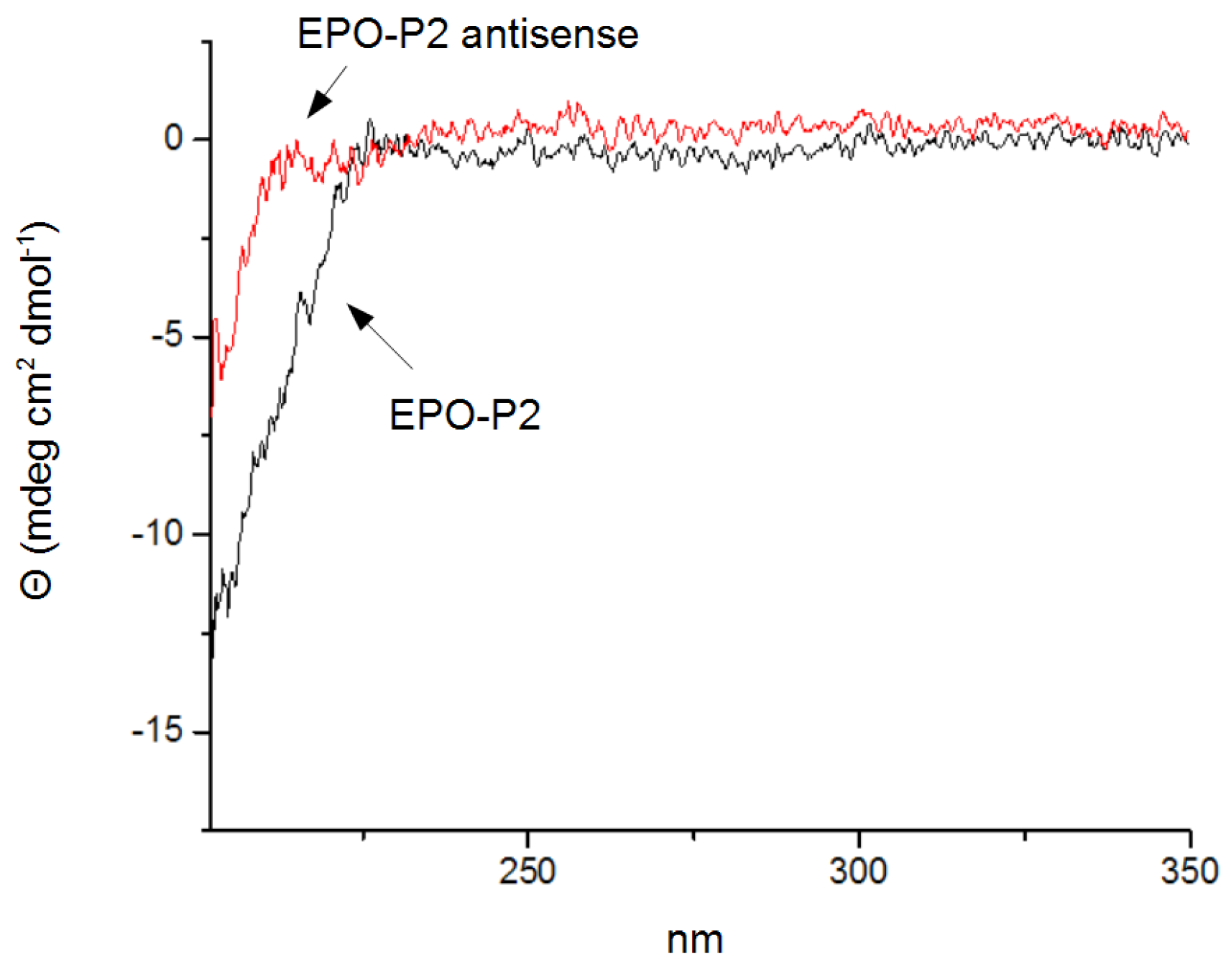
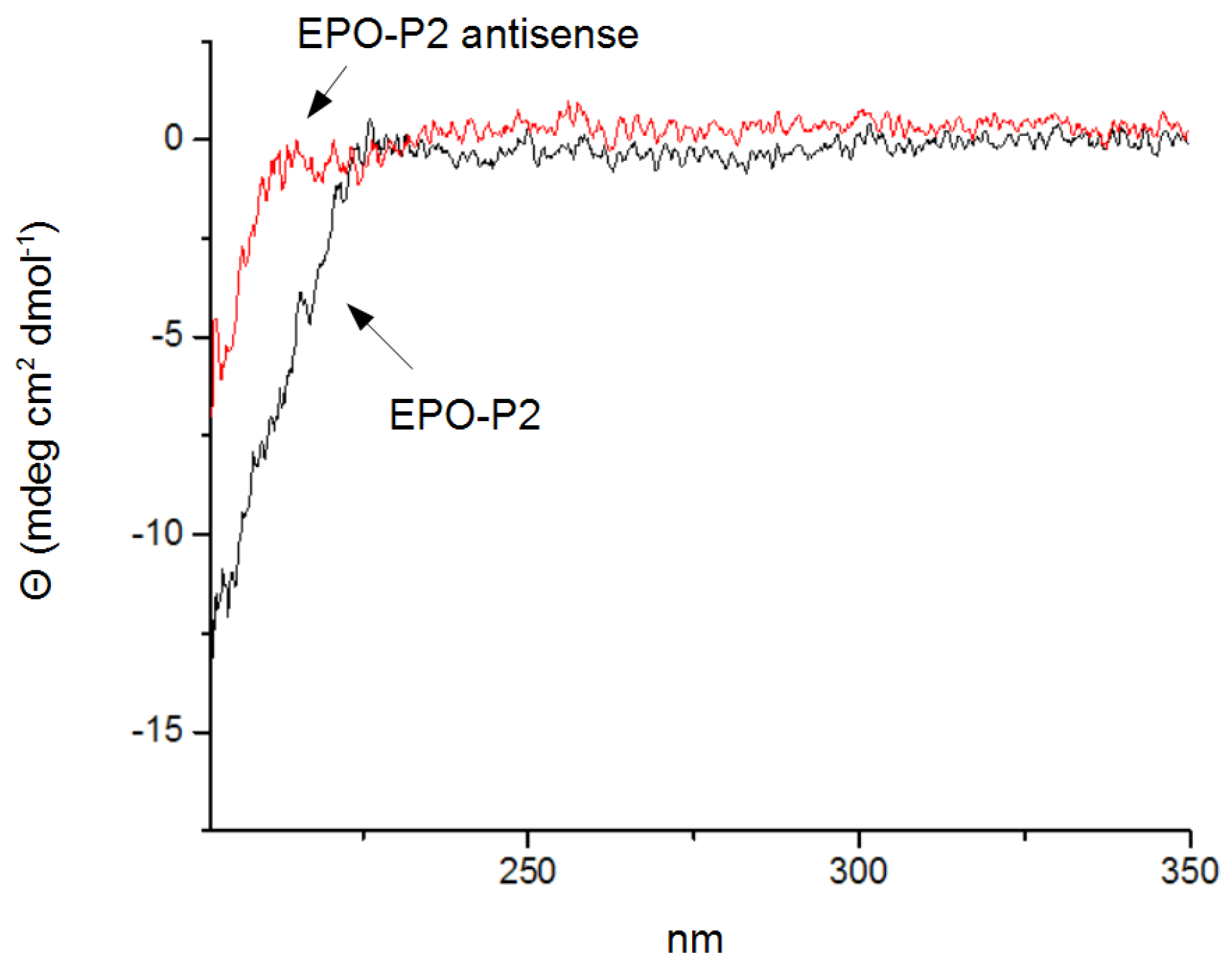
3.4. Circular Dichroism Spectroscopy
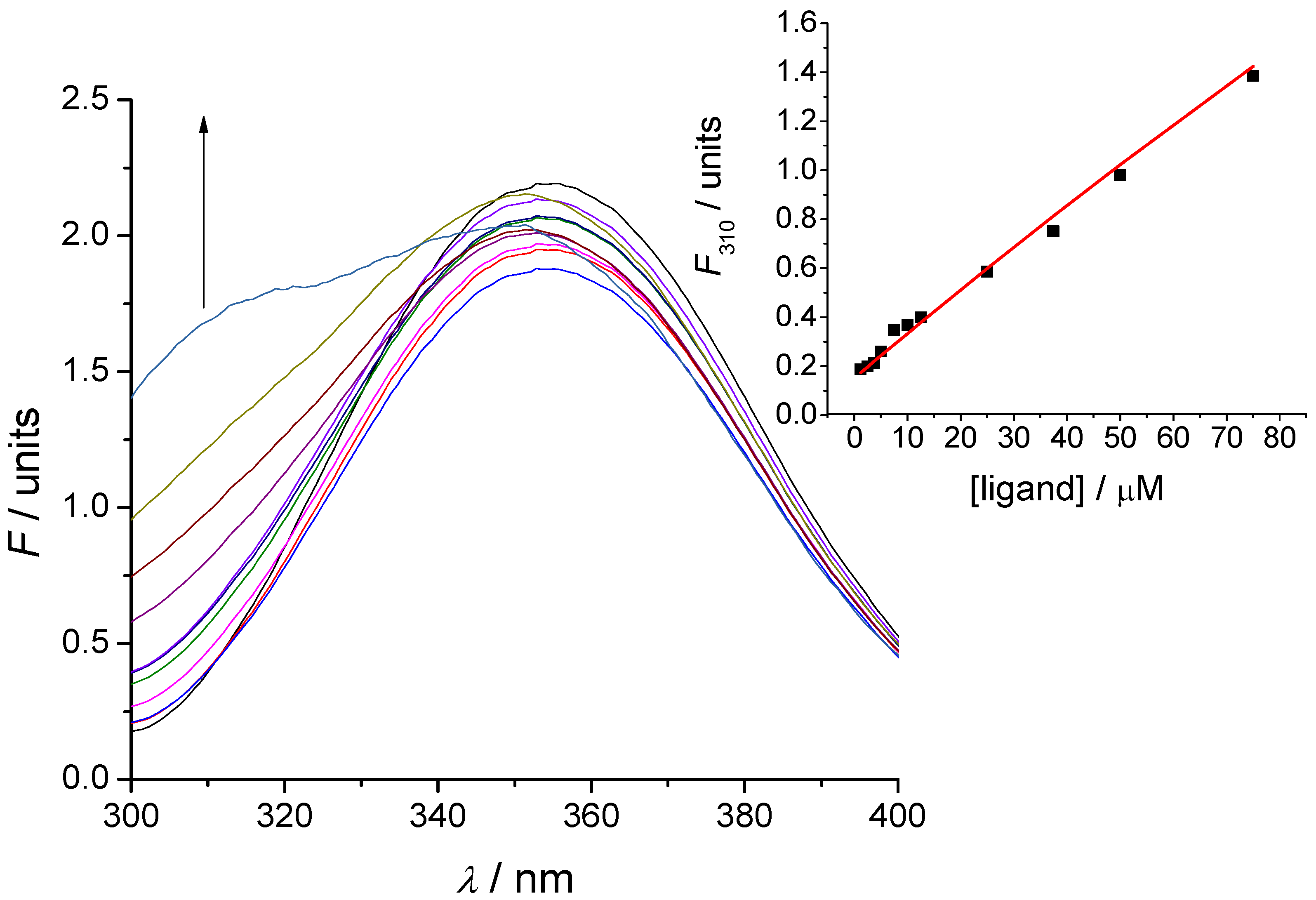
3.5. Tryptophan Fluorescence Spectroscopy
3.6. Microscale Thermophoresis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heal, J.R.; Roberts, G.W.; Raynes, J.G.; Bhakoo, A.; Miller, A.D. Specific interactions between sense and complementary peptides: The basis for the proteomic code. ChemBioChem 2002, 3, 136–151. [Google Scholar]
- Biro, J.C. The proteonomic code: A molecular recognition code for proteins. Theor. Biol. Med. Model. 2007, 4, 1–45. [Google Scholar] [CrossRef]
- Blalock, J.E. Genetic origin of protein shape and interaction rules. Nat. Med. 1995, 1, 876–878. [Google Scholar] [CrossRef]
- Houra, K.; Turčić, P.; Gabričević, M.; Weitner, T.; Konjevoda, P.; Štambuk, N. Interaction of α-melanocortin and its pentapeptide antisense LVKAT: Effects on hepatoprotection in male CBA mice. Molecules 2011, 16, 7331–7343. [Google Scholar] [CrossRef]
- McGuire, K.L.; Holmes, D.S. Role of complementary proteins in autoimmunity: An old idea re-emerges with new twists. Trends Immunol. 2005, 26, 367–372. [Google Scholar] [CrossRef]
- tambuk, N.; Konjevoda, P.; Boban-Blagaić, A.; Pokrić, B. Molecular recognition theory of the complementary (antisense) peptide interactions. Theory Biosci. 2005, 123, 265–275. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Peptide self-aggregation and peptide complementarity as bases for the evolution of peptide receptors: A review. J. Mol. Recognit. 2005, 18, 40–49. [Google Scholar]
- Molineux, G.; Sinclair, A.M. Biology of erythropoietin. In Erythropoietins, Erythropoietic Factors and Erythropoiesis; Elliott, S.G., Foote, M.A., Molineux, G., Eds.; Birkhäuser Verlag: Basel, Switzerland, 2009; pp. 41–60. [Google Scholar]
- Walsh, G. Biopharmaceuticals: Biochemistry and Biotechnology, 2nd ed.; Wiley: Chichester, UK, 2003; pp. 264–272. [Google Scholar]
- Reichel, C.; Gmeiner, G. Erythropoietin and analogs. In Doping in Sports; Thieme, D., Hemmersbach, P., Eds.; Springer-Verlag: Heildeberg, Germany, 2010; pp. 251–294. [Google Scholar]
- Fibi, M.R.; Stüber, W.; Hintz-Obertreis, P.; Lüben, G.; Krumwieh, D.; Siebold, B.; Zettlmeissl, G.; Küpper, H.A. Evidence for the location of the receptor-binding site of human erythropoietin at the carboxyl-terminal domain. Blood 1991, 77, 1203–1210. [Google Scholar]
- Brown, F.; Doughan, G.; Hoey, E.M.; Martin, S.J.; Rima, B.K.; Trudgett, A. Vaccine Design; Wiley: Chichester, UK, 1993; pp. 33–44. [Google Scholar]
- Hopp, T.P.; Woods, K.R. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3824–3828. [Google Scholar]
- Jameson, B.A.; Wolf, H. The antigenic index: A novel algorithm for predicting antigenic determinants. Comput. Appl. Biosci. 1988, 4, 181–186. [Google Scholar]
- Cosic, I. The Resonant Recognition Model of Macromolecular Bioactivity: Theory and Applications; Birkhäuser: Basel, Switzerland, 1997; pp. 1–87. [Google Scholar]
- Cosic, I.; Nesic, D. Prediction of “hot spots” in SV40 enhancer and relation with experimental data. Eur. J. Biochem. 1987, 170, 247–252. [Google Scholar] [CrossRef]
- Zelterman, D. Applied Linear Models with SAS; Cambridge University Press: Cambridge, UK, 2010; pp. 75–119. [Google Scholar]
- Biro, J.C. Discovery of proteomic code with mRNA assisted protein folding. Int. J. Mol. Sci. 2008, 9, 2424–2446. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S. Amino acid pairing. J. Theor. Biol. 1982, 94, 885–894. [Google Scholar] [CrossRef]
- Blalock, J.E.; Bost, K.L. Binding of peptides that are specified by complementary RNAs. Biochem. J. 1986, 234, 679–683. [Google Scholar]
- Siemion, I.Z.; Stefanowicz, P. Periodical changes of amino acid reactivity within the genetic code. Biosystems 1992, 27, 77–84. [Google Scholar] [CrossRef]
- Siemion, I.Z.; Cebrat, M.; Kluczyk, A. The problem of amino acid complementarity and antisense peptides. Curr. Protein Peptide Sci. 2004, 5, 507–527. [Google Scholar] [CrossRef]
- Štambuk, N. On the genetic origin of complementary protein coding. Croat. Chem. Acta. 1998, 71, 573–589. [Google Scholar]
- Houra, K.; Štambuk, N.; Konjevoda, P.; Boban-Blagaić, A.; Bruketa, T.; Pokrić, B. α-Melanotropin peptide: Structure and ligand–receptor recognition. Croat. Chem. Acta 2006, 79, 379–383. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of equilibrium constants from multiwavelength spectroscopic data-I mathematical considerations. Talanta. 1985, 32, 95–101. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of equilibrium constants from multiwavelength spectroscopic data-II: SPECFIT: Two user-friendly programs in basic and standard FORTRAN 77. Talanta. 1985, 32, 257–264. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of equilibrium constants from multiwavelength spectroscopic data-IV Model-free least-squares refinement by use of evolving factor analysis. Talanta 1986, 33, 943–951. [Google Scholar] [CrossRef]
- Maeder, M.; Neuhold, Y.-M. Practical Data Analysis; Elsevier B.V.: Amsterdam, The Netherlands, 2007; pp. 214–217. [Google Scholar]
- Baaske, P.; Wienken, C.J.; Reineck, P.; Duhr, S.; Braun, D. Optical thermophoresis quantifies buffer dependence of aptamer binding. Angew. Chem. Int. Ed. 2010, 49, 1–5. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef]
- Bost, K.L.; Smith, E.M.; Blalock, J.E. Similarity between the corticotropin (ACTH) receptor and a peptide encoded by an RNA that is complementary to ACTH mRNA. Proc. Natl. Acad. Sci. USA 1985, 82, 1372–1375. [Google Scholar] [CrossRef]
- Fassina, G.; Cassini, G. Design and recognition properties of a hydropathically complementary peptide to human interleukin 1β. Biochem. J. 1992, 282, 773–779. [Google Scholar]
- Martínez-Ceron, M.C.; Targovnik, A.M.; Urtasun, N.; Cascone, O.; Miranda, M.V.; Camperi, S.A. Recombinant protein purification using complementary peptides as affinity tags. Biotechnology 2012, 29, 206–210. [Google Scholar]
- Heal, J.R.; Roberts, G.W.; Christie, G.; Miller, A.D. Inhibition of β-amyloid aggregation and neurotoxicity by complementary (antisense) peptides. ChemBioChem 2002, 3, 86–92. [Google Scholar] [CrossRef]
- Heal, J.R.; Bino, S.; Roberts, G.W.; Raynes, J.G.; Miller, A.D. Mechanistic investigation into complementary (antisense) peptide mini-receptor inhibitors of cytokine interleukin-1. ChemBiochem 2002, 3, 76–85. [Google Scholar] [CrossRef]
- Root-Bernstein, R.S.; Westall, F.C. Bovine pineal antireproductive tripeptide binds to luteinizing hormone-releasing hormone: A model for peptide modulation by sequence specific peptide interactions? Brain Res. Bull. 1986, 17, 519–528. [Google Scholar] [CrossRef]
- Loo, J.L. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997, 16, 1–23. [Google Scholar] [CrossRef]
- Bhakoo, A.; Raynes, J.G.; Heal, J.R.; Keller, M.K.; Miller, A.D. De novo design of complementary (antisense) peptide mini-receptor inhibitor of interleukin 18 (IL-18). Mol. Immunol. 2004, 41, 1217–1224. [Google Scholar] [CrossRef]
- Seidel, S.A.; Dijkman, P.M.; Lea, W.A.; van den Bogaart, G.; Jerabek-Willemsen, M.; Lazic, A.; Joseph, J.S.; Srinivasan, P.; Baaske, P.; Simeonov, A.; et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods 2013, 59, 301–315. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; André, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. Microscale thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014. [Google Scholar] [CrossRef]
- Seidel, S.A.; Wienken, C.J.; Geissler, S.; Jerabek-Willemsen, M.; Duhr, S.; Reiter, A.; Trauner, D.; Braun, D.; Baaske, P. Label-free microscale thermophoresis discriminates sites and affinity of protein-ligand binding. Angew. Chem. Int. Ed. 2012, 51, 10656–10659. [Google Scholar] [CrossRef]
- Parker, J.L.; Newstead, S. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature 2014, 507, 68–72. [Google Scholar]
- Tetin, S.Y.; Hazlett, T.L. Optical spectroscopy in studies of antibody-hapten interactions. Methods 2000, 20, 341–361. [Google Scholar] [CrossRef]
- Cheetham, J.C.; Smith, D.M.; Aoki, K.H.; Stevenson, J.L.; Hoeffel, T.J.; Syed, R.S.; Egrie, J.; Harvey, T.S. NMR structure of human erythropoietin and a comparison with its receptor bound conformation. Nat. Struct. Biol. 1998, 5, 861–866. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; the UGENE team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- STATISTICA Version 8.0. Available online: http://www.statsoft.com (accessed on 23 December 2013).
- Schleker, S.; Ananthasubramanian, S.; Klein-Seetharaman, J.; Ganapathiraju, M.K. Prediction of intra- and inter-species protein–protein interactions facilitating systems biology studies. In Protein-Protein Interactions in Drug Discovery; Dömling, A., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 21–54. [Google Scholar]
- Root-Bernstein, R.S.; Dillon, P.F. Small molecule complementarity as a source of novel pharmaceutical agents and combination therapies. Curr. Pharm. Des. 2008, 14, 55–62. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Štambuk, N.; Manojlović, Z.; Turčić, P.; Martinić, R.; Konjevoda, P.; Weitner, T.; Wardega, P.; Gabričević, M. A Simple Three-Step Method for Design and Affinity Testing of New Antisense Peptides: An Example of Erythropoietin. Int. J. Mol. Sci. 2014, 15, 9209-9223. https://doi.org/10.3390/ijms15069209
Štambuk N, Manojlović Z, Turčić P, Martinić R, Konjevoda P, Weitner T, Wardega P, Gabričević M. A Simple Three-Step Method for Design and Affinity Testing of New Antisense Peptides: An Example of Erythropoietin. International Journal of Molecular Sciences. 2014; 15(6):9209-9223. https://doi.org/10.3390/ijms15069209
Chicago/Turabian StyleŠtambuk, Nikola, Zoran Manojlović, Petra Turčić, Roko Martinić, Paško Konjevoda, Tin Weitner, Piotr Wardega, and Mario Gabričević. 2014. "A Simple Three-Step Method for Design and Affinity Testing of New Antisense Peptides: An Example of Erythropoietin" International Journal of Molecular Sciences 15, no. 6: 9209-9223. https://doi.org/10.3390/ijms15069209