NOD-Like Receptors in Intestinal Homeostasis and Epithelial Tissue Repair

Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Intestinal Epithelial Barrier
3. Process of Intestinal Epithelial Tissue Repair
4. Mechanisms of Intestinal Epithelial Tissue Repair
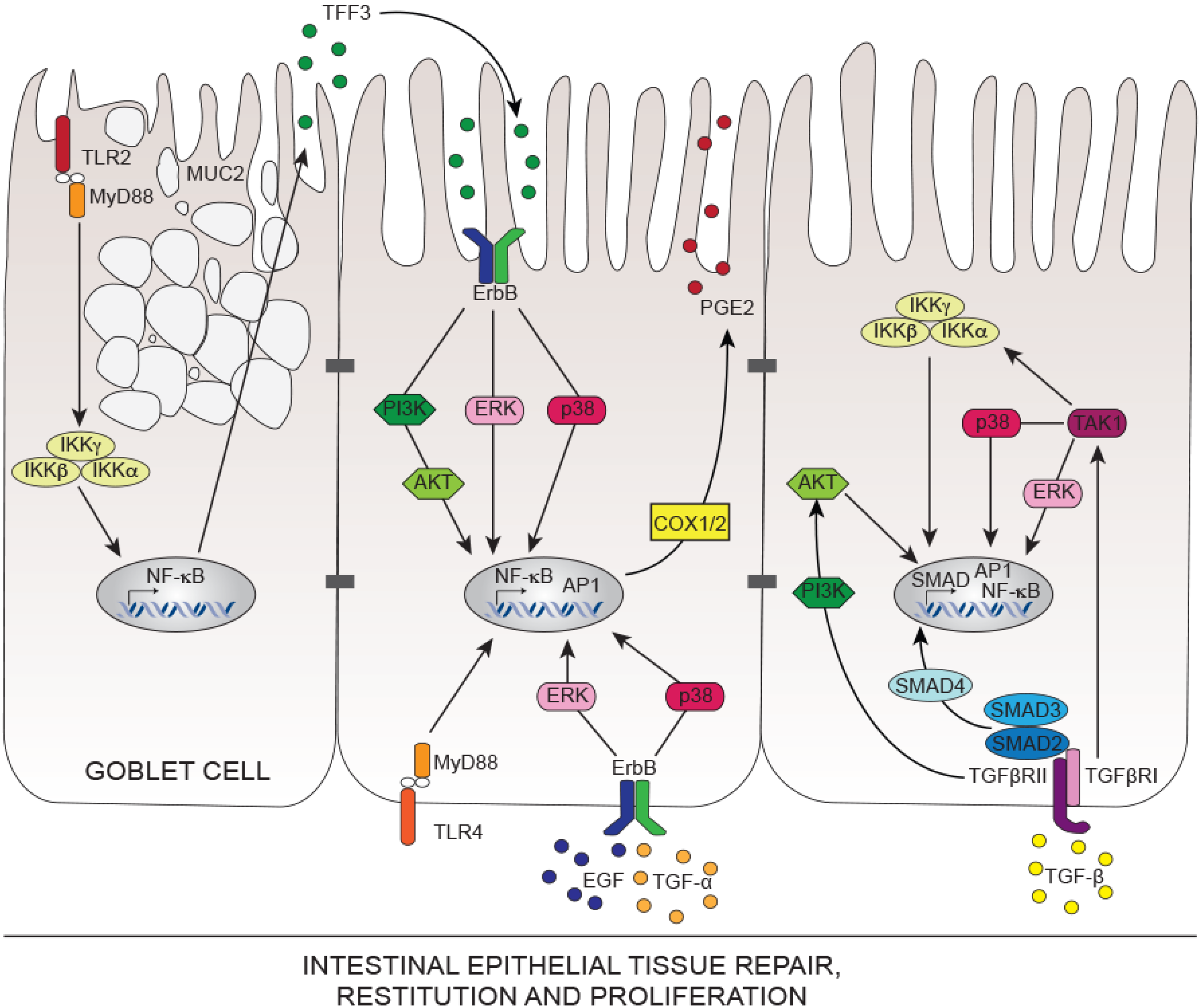
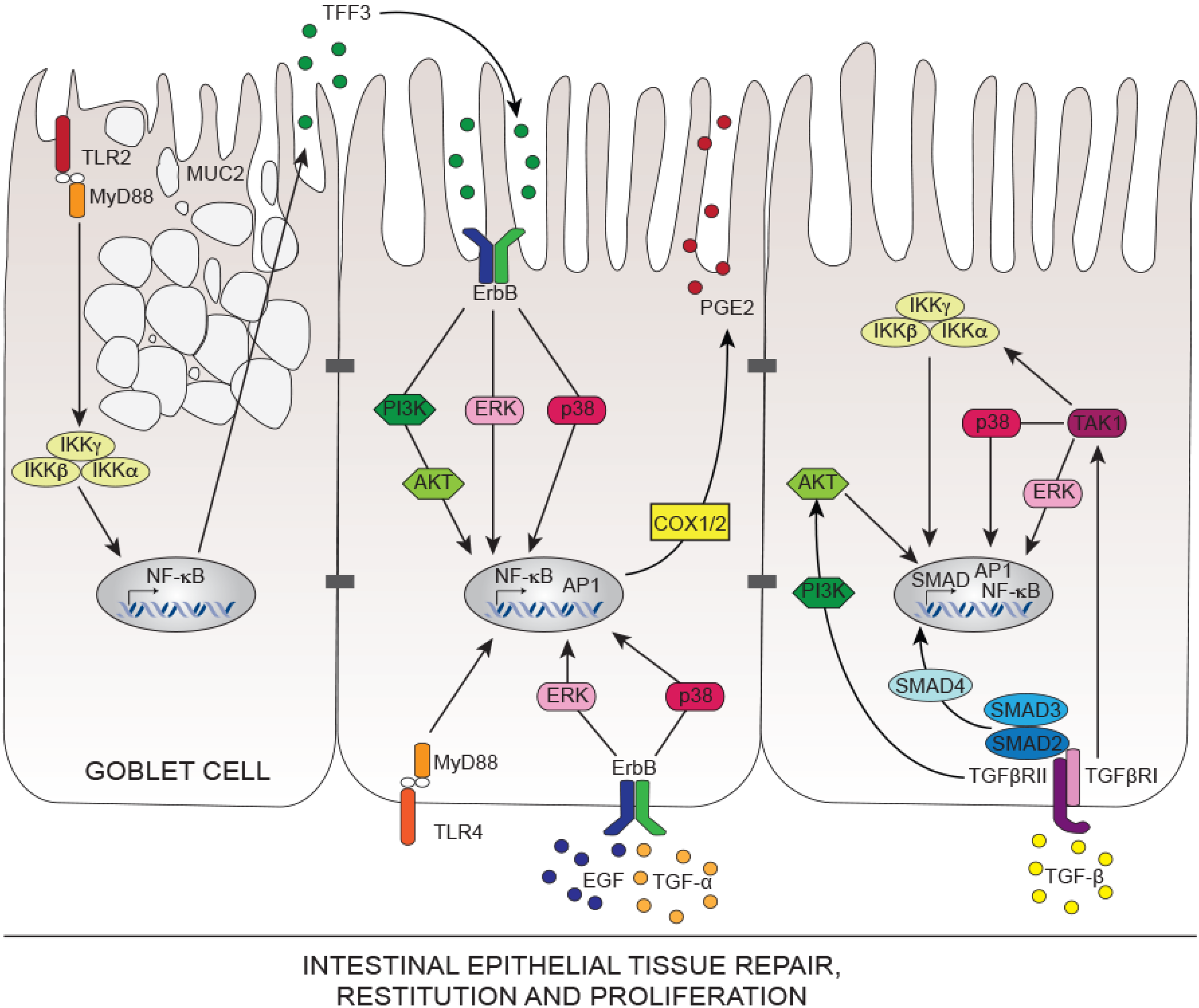
5. Innate Immunity Controlling Intestinal Tissue Repair
5.1. Toll-Like Receptors TLRs in Intestinal Tissue Repair
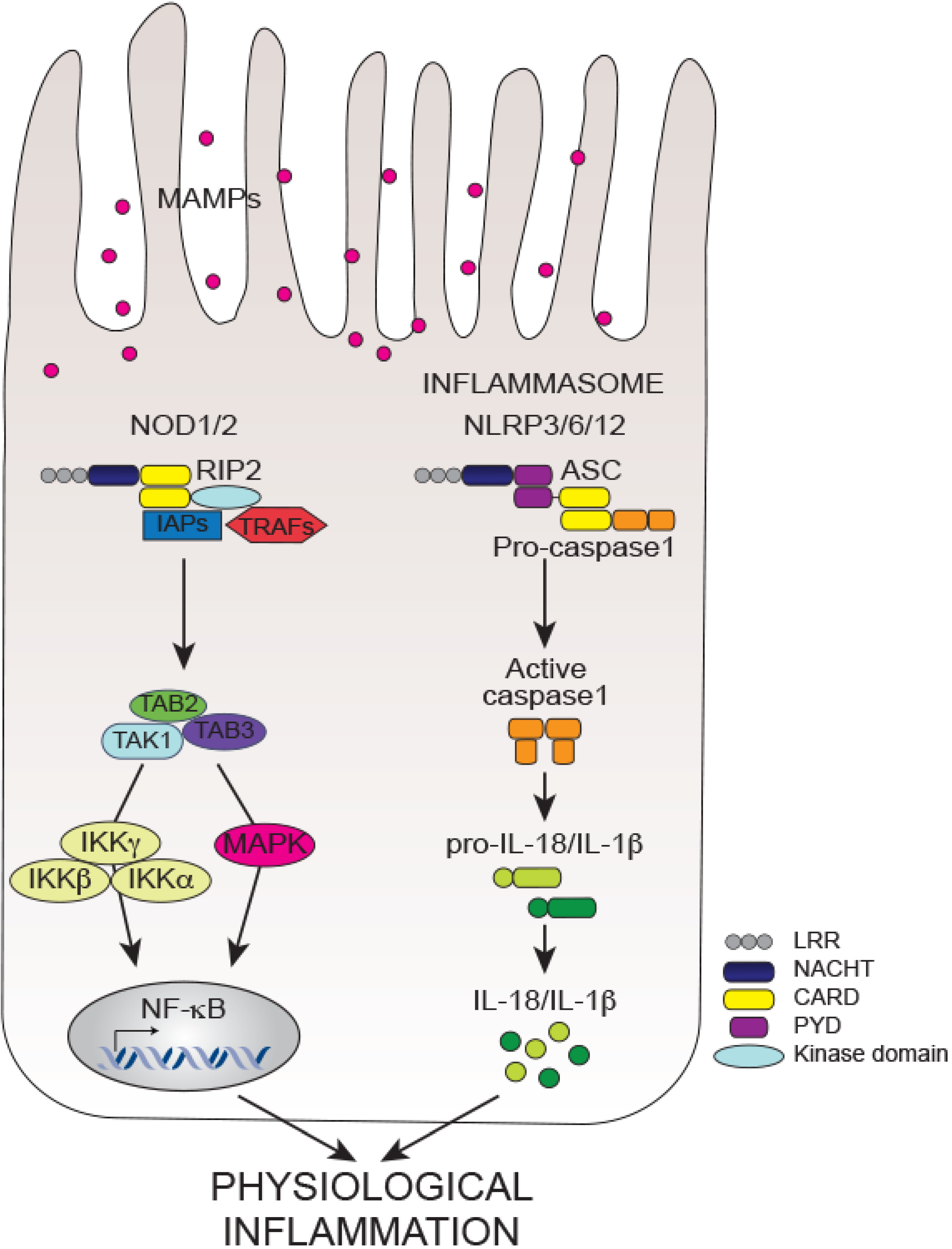
5.2. NOD-Like Receptors (NLRs) in Intestinal Homeostasis, Inflammation and Tissue Repair
5.2.1. Non-Inflammasome NLRs
5.2.2. Inflammasome NLRs
6. Conclusions
Abbreviations
| IECs | intestinal epithelial cells |
| IBD | inflammatory bowel diseases |
| NLRs | nucleotide-binding and oligomerization domain-like receptors |
| TLRs | toll-like receptors |
| CD | Crohn’s disease |
| UC | ulcerative colitis |
| TH | T helper |
| TGF-β | transforming-growth factor-β |
| Foxp3 | forkhead box P3 |
| STAT3 | signal transducer and activator of transcription 3 |
| HNF4A | hepatocyte nuclear factor 4 alpha |
| ITLN1 | intelectin 1 |
| MUC19 | mucin 19 |
| NOD2 | nucleotide-binding oligomerization domain containing 2 |
| CARD9 | caspase recruitment domain family, member 9 |
| RIPK2 | receptor interacting protein kinase 2 |
| NLRP3 | NLR family, pyrin domain containing 3 |
| ATG16L1 | autophagy related 16-like 1 |
| IRGM | immunity-related GTPase family, M |
| LRRK2 | leucine-rich repeat kinase 2 |
| IL23R | interleukin 23 receptor |
| IL10R | interleukin 10 receptor |
| JAK2 | janus kinase 2 |
| MUC2 | mucin 2 |
| Lgr5 | leucine-rich repeat-containing G-protein coupled receptor 5 |
| TA | transit-amplifying |
| ISCs | intestinal stem cells |
| GTPases | guanine triphosphatases |
| RHO | rhodopsin |
| CDC42 | cell division cycle 42 |
| CXCL | chemokine (C-X-C motif) ligand |
| HBD2 | human β-defensin 2 |
| CCL2 | chemokine (C-C motif) ligand 2 |
| CCR6 | chemokine (C-C motif) receptor 6 |
| DSS | dextran sulfate sodium |
| MMP7 | Matrix metalloproteinase-7 |
| TFF | trefoil factor |
| EGFR | epidermal growth factor receptor |
| EGF | epidermal growth factor |
| ErbB | v-erb-b2 avian erythroblastic leukemia viral oncogene homolog |
| MAPK | mitogen activated protein kinase |
| PI3K | phosphoinositol 3-kinase |
| TNF-α | tumor necrosis factor alpha |
| NF-κB | nuclear factor-kappa B |
| ΙΚΚα | IκB kinase alpha |
| ΙΚΚβ | IκB kinase beta |
| ΙΚΚγ | IκB kinase gamma |
| NEMO | NF-κB Essential Modulator |
| HGF | hepatocyte growth factor |
| VEGF | vascular endothelial cell growth factor |
| GM-CSF | granulocyte macrophage colony stimulating factor |
| KGF | keratinocyte growth factor |
| PDGF | platelet-derived growth factor |
| IFN | interferon |
| ERK | extracellular signal regulated-kinases |
| TNBS | trinitrobenzene sulphonic acid |
| COX1/2 | cyclooxygenase 1/2 |
| SMAD | Sma- And Mad-Related Protein |
| TGFβRII | TGFβ receptor II |
| TAK1 | TGF-β activated kinase 1 |
| Wnt5a | wingless-type MMTV integration site family, member 5A |
| PGE2 | Prostaglandin-E2 |
| MyD88 | Myeloid Differentiation Primary Response 88 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| HES1 | hairy enhancer of split 1 |
| LRR | leucine-rich repeats |
| NACHT | nucleotide-binding and oligomerization domain |
| BIR | baculovirus inhibitor of apoptosis repeat |
| CARD | caspase activation and recruitment domain |
| PYD | pyrin domain |
| MAMPs | microbial-associated molecular patterns |
| DAMPs | danger-associated molecular patterns |
| DAP | d-glutamyl-meso-diaminopimelic acid |
| MDP | muramyl dipeptide |
| XIAP | X-linked inhibitor of apoptosis |
| cIAP | cellular inhibitor of apoptosis protein |
| TRAF | TNFR-associated factor |
| TAB2-TAB3 | TAK-1-binding protein 2 and 3 |
| RNAi | ribonucleic acid interference |
| FRMPD2 | FERM and PDZ domain protein-containing 2 |
| ERBIN | ERBB2-interacting protein |
| ASC | apoptosis-associated speck-like protein |
| IL18RAP | Interleukin 18 Receptor Accessory Protein |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef]
- Cho, J.H. The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 2008, 8, 458–466. [Google Scholar] [CrossRef]
- Long, M.D.; Hutfless, S.; Kappelman, M.D.; Khalili, H.; Kaplan, G.G.; Bernstein, C.N.; Colombel, J.F.; Gower-Rousseau, C.; Herrinton, L.; Velayos, F.; et al. Challenges in designing a national surveillance program for inflammatory bowel disease in the united states. Inflamm. Bowel Dis. 2014, 20, 398–415. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Garrett, W.S.; Gordon, J.I.; Glimcher, L.H. Homeostasis and inflammation in the intestine. Cell 2010, 140, 859–870. [Google Scholar] [CrossRef]
- Lochner, M.; Ohnmacht, C.; Presley, L.; Bruhns, P.; Si-Tahar, M.; Sawa, S.; Eberl, G. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of rorgamma T and LTI cells. J. Exp. Med. 2011, 208, 125–134. [Google Scholar] [CrossRef]
- Olszak, T.; An, D.; Zeissig, S.; Vera, M.P.; Richter, J.; Franke, A.; Glickman, J.N.; Siebert, R.; Baron, R.M.; Kasper, D.L.; et al. Microbial exposure during early life has persistent effects on natural killer t cell function. Science 2012, 336, 489–493. [Google Scholar] [CrossRef]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar]
- Casellas, F.; Borruel, N.; Papo, M.; Guarner, F.; Antolin, M.; Videla, S.; Malagelada, J.R. Antiinflammatory effects of enterically coated amoxicillin-clavulanic acid in active ulcerative colitis. Inflamm. Bowel Dis. 1998, 4, 1–5. [Google Scholar] [CrossRef]
- Macpherson, A.; Khoo, U.Y.; Forgacs, I.; Philpott-Howard, J.; Bjarnason, I. Mucosal antibodies in inflammatory bowel disease are directed against intestinal bacteria. Gut 1996, 38, 365–375. [Google Scholar] [CrossRef]
- Pirzer, U.; Schonhaar, A.; Fleischer, B.; Hermann, E.; Meyer zum Buschenfelde, K.H. Reactivity of infiltrating T lymphocytes with microbial antigens in crohn’s disease. Lancet 1991, 338, 1238–1239. [Google Scholar] [CrossRef]
- Frank, D.N.; St. Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar]
- Dicksved, J.; Halfvarson, J.; Rosenquist, M.; Jarnerot, G.; Tysk, C.; Apajalahti, J.; Engstrand, L.; Jansson, J.K. Molecular analysis of the gut microbiota of identical twins with crohn’s disease. ISME J. 2008, 2, 716–727. [Google Scholar] [CrossRef]
- Sartor, R.B. Microbial influences in inflammatory bowel diseases. Gastroenterology 2008, 134, 577–594. [Google Scholar] [CrossRef]
- Guarner, F. What is the role of the enteric commensal flora in IBD? Inflamm. Bowel Dis. 2008, 14, S83–S84. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjoberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immun. 2010, 11, 76–83. [Google Scholar] [CrossRef]
- Scanlan, P.D.; Shanahan, F.; O’Mahony, C.; Marchesi, J.R. Culture-independent analyses of temporal variation of the dominant fecal microbiota and targeted bacterial subgroups in crohn’s disease. J. Clin. Microbiol. 2006, 44, 3980–3988. [Google Scholar]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Hedderich, J.; May, S.; Lu, T.; Schuldt, D.; Nikolaus, S.; Rosenstiel, P.; Krawczak, M.; et al. Replication of signals from recent studies of crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat. Genet. 2008, 40, 713–715. [Google Scholar] [CrossRef]
- Lepage, P.; Hasler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Dore, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lecuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; de Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper t cell responses. Immunity 2009, 31, 677–689. [Google Scholar]
- Wu, H.J.; Ivanov, II; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef]
- Ivanov, II; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef]
- Janse, M.; Lamberts, L.E.; Franke, L.; Raychaudhuri, S.; Ellinghaus, E.; Muri Boberg, K.; Melum, E.; Folseraas, T.; Schrumpf, E.; Bergquist, A.; et al. Three ulcerative colitis susceptibility loci are associated with primary sclerosing cholangitis and indicate a role for IL2, REL, and CARD9. Hepatology 2011, 53, 1977–1985. [Google Scholar] [CrossRef]
- Danoy, P.; Pryce, K.; Hadler, J.; Bradbury, L.A.; Farrar, C.; Pointon, J.; Australo-Anglo-American Spondyloarthritis Consortium; Ward, M.; Weisman, M.; Reveille, J.D.; et al. Association of variants at 1q32 and STAT3 with ankylosing spondylitis suggests genetic overlap with crohn’s disease. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef] [Green Version]
- Consortium, U.I.G.; Barrett, J.C.; Lee, J.C.; Lees, C.W.; Prescott, N.J.; Anderson, C.A.; Phillips, A.; Wesley, E.; Parnell, K.; Zhang, H.; et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4a region. Nat. Genet. 2009, 41, 1330–1334. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Beaudoin, M.; Goyette, P.; Boucher, G.; Lo, K.S.; Rivas, M.A.; Stevens, C.; Alikashani, A.; Ladouceur, M.; Ellinghaus, D.; Torkvist, L.; et al. Deep resequencing of gwas loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [Green Version]
- Villani, A.C.; Lemire, M.; Fortin, G.; Louis, E.; Silverberg, M.S.; Collette, C.; Baba, N.; Libioulle, C.; Belaiche, J.; Bitton, A.; et al. Common variants in the nlrp3 region contribute to crohn’s disease susceptibility. Nat. Genet. 2009, 41, 71–76. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; de La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Duraes, C.; Machado, J.C.; Portela, F.; Rodrigues, S.; Lago, P.; Cravo, M.; Ministro, P.; Marques, M.; Cremers, I.; Freitas, J.; et al. Phenotype-genotype profiles in crohn’s disease predicted by genetic markers in autophagy-related genes (GOIA study II). Inflamm. Bowel Dis. 2013, 19, 230–239. [Google Scholar] [CrossRef]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar]
- Tremelling, M.; Cummings, F.; Fisher, S.A.; Mansfield, J.; Gwilliam, R.; Keniry, A.; Nimmo, E.R.; Drummond, H.; Onnie, C.M.; Prescott, N.J.; et al. IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology 2007, 132, 1657–1664. [Google Scholar] [CrossRef]
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schaffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef]
- Anderson, C.A.; Massey, D.C.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Fisher, S.A.; Gwilliam, R.; Jacob, J.; Nimmo, E.R.; Drummond, H.; et al. Investigation of crohn’s disease risk loci in ulcerative colitis further defines their molecular relationship. Gastroenterology 2009, 136, 523–529. [Google Scholar] [CrossRef]
- Laukoetter, M.G.; Bruewer, M.; Nusrat, A. Regulation of the intestinal epithelial barrier by the apical junctional complex. Curr. Opin. Gastroenterol. 2006, 22, 85–89. [Google Scholar] [CrossRef]
- Muniz, L.R.; Knosp, C.; Yeretssian, G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front. Immun. 2012, 3, 310. [Google Scholar]
- Sturm, A.; Dignass, A.U. Epithelial restitution and wound healing in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 348–353. [Google Scholar] [CrossRef]
- Harris, G.; KuoLee, R.; Chen, W. Role of toll-like receptors in health and diseases of gastrointestinal tract. World J. Gastroenterol. 2006, 12, 2149–2160. [Google Scholar]
- Iizuka, M.; Konno, S. Wound healing of intestinal epithelial cells. World J. Gastroenterol. 2011, 17, 2161–2171. [Google Scholar] [CrossRef]
- Yeretssian, G. Effector functions of NLRs in the intestine: Innate sensing, cell death, and disease. Immun. Res. 2012, 54, 25–36. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Allez, M.; Brimnes, J.; Dotan, I.; Mayer, L. Expansion of CD8+ T cells with regulatory function after interaction with intestinal epithelial cells. Gastroenterology 2002, 123, 1516–1526. [Google Scholar] [CrossRef]
- Rabinowitz, K.; Mayer, L. Working out mechanisms of controlled/physiologic inflammation in the GI tract. Immun. Res. 2012, 54, 14–24. [Google Scholar] [CrossRef]
- Roda, G.; Jianyu, X.; Park, M.S.; Demarte, L.; Hovhannisyan, Z.; Couri, R.; Stanners, C.P.; Yeretssian, G.; Mayer, L. Characterizing CEACAM5 interaction with CD8α and CD1D in intestinal homeostasis. Mucosal Immunol. 2013, 7, 615–624. [Google Scholar]
- Johansson, M.E.; Sjovall, H.; Hansson, G.C. The gastrointestinal mucus system in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 352–361. [Google Scholar] [CrossRef]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and function of the polymeric mucins in airways mucus. Ann. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two MUC2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar]
- Shan, M.; Gentile, M.; Yeiser, J.R.; Walland, A.C.; Bornstein, V.U.; Chen, K.; He, B.; Cassis, L.; Bigas, A.; Cols, M.; et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 2013, 342, 447–453. [Google Scholar] [CrossRef]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Stange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for LGR5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef]
- Watson, A.J.; Hughes, K.R. Tnf-α-induced intestinal epithelial cell shedding: Implications for intestinal barrier function. Ann. N. Y. Acad. Sci. 2012, 1258, 1–8. [Google Scholar] [CrossRef]
- Eisenhoffer, G.T.; Loftus, P.D.; Yoshigi, M.; Otsuna, H.; Chien, C.B.; Morcos, P.A.; Rosenblatt, J. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 2012, 484, 546–549. [Google Scholar] [CrossRef]
- Marinari, E.; Mehonic, A.; Curran, S.; Gale, J.; Duke, T.; Baum, B. Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature 2012, 484, 542–545. [Google Scholar] [CrossRef]
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef]
- Barker, N.; Bartfeld, S.; Clevers, H. Tissue-resident adult stem cell populations of rapidly self-renewing organs. Cell Stem Cell 2010, 7, 656–670. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene LGR5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single LGR5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Clevers, H. Stem cells: A unifying theory for the crypt. Nature 2013, 495, 53–54. [Google Scholar] [CrossRef]
- Barker, N.; van Oudenaarden, A.; Clevers, H. Identifying the stem cell of the intestinal crypt: Strategies and pitfalls. Cell Stem Cell 2012, 11, 452–460. [Google Scholar] [CrossRef]
- Buczacki, S.J.; Zecchini, H.I.; Nicholson, A.M.; Russell, R.; Vermeulen, L.; Kemp, R.; Winton, D.J. Intestinal label-retaining cells are secretory precursors expressing LGR5. Nature 2013, 495, 65–69. [Google Scholar] [CrossRef]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism and applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef]
- Huch, M.; Dorrell, C.; Boj, S.F.; van es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single LGR5+ liver stem cells induced by wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef]
- Yui, S.; Nakamura, T.; Sato, T.; Nemoto, Y.; Mizutani, T.; Zheng, X.; Ichinose, S.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; et al. Functional engraftment of colon epithelium expanded in vitro from a single adult LGR5+ stem cell. Nat. Med. 2012, 18, 618–623. [Google Scholar]
- Fordham, R.P.; Yui, S.; Hannan, N.R.; Soendergaard, C.; Madgwick, A.; Schweiger, P.J.; Nielsen, O.H.; Vallier, L.; Pedersen, R.A.; Nakamura, T.; et al. Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell 2013, 13, 734–744. [Google Scholar] [CrossRef]
- Owens, B.M.; Simmons, A. Intestinal stromal cells in mucosal immunity and homeostasis. Mucosal Immunol. 2013, 6, 224–234. [Google Scholar] [CrossRef]
- Banan, A.; Choudhary, S.; Zhang, Y.; Fields, J.Z.; Keshavarzian, A. Oxidant-induced intestinal barrier disruption and its prevention by growth factors in a human colonic cell line: Role of the microtubule cytoskeleton. Free Radic. Biol. Med. 2000, 28, 727–738. [Google Scholar] [CrossRef]
- Dean, P.; Kenny, B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol. Microbiol. 2004, 54, 665–675. [Google Scholar] [CrossRef]
- Moore, R.; Carlson, S.; Madara, J.L. Rapid barrier restitution in an in vitro model of intestinal epithelial injury. Lab. Investig. 1989, 60, 237–244. [Google Scholar]
- McCormack, S.A.; Viar, M.J.; Johnson, L.R. Migration of IEC-6 cells: A model for mucosal healing. Am. J. Physiol. 1992, 263, G426–G435. [Google Scholar]
- Nusrat, A.; Delp, C.; Madara, J.L. Intestinal epithelial restitution. Characterization of a cell culture model and mapping of cytoskeletal elements in migrating cells. J. Clin. Investig. 1992, 89, 1501–1511. [Google Scholar] [CrossRef]
- Karrasch, T.; Jobin, C. Wound healing responses at the gastrointestinal epithelium: A close look at novel regulatory factors and investigative approaches. Z. Gastroenterol. 2009, 47, 1221–1229. [Google Scholar] [CrossRef]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33, 891–895. [Google Scholar]
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Ann. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef]
- Cau, J.; Hall, A. CDC42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J. Cell Sci. 2005, 118, 2579–2587. [Google Scholar] [CrossRef]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef]
- Moyer, R.A.; Wendt, M.K.; Johanesen, P.A.; Turner, J.R.; Dwinell, M.B. Rho activation regulates CXCL12 chemokine stimulated actin rearrangement and restitution in model intestinal epithelia. Lab. Investig. 2007, 87, 807–817. [Google Scholar] [CrossRef]
- Vongsa, R.A.; Zimmerman, N.P.; Dwinell, M.B. CCR6 regulation of the actin cytoskeleton orchestrates human β defensin-2 and CCL20-mediated restitution of colonic epithelial cells. J. Biol. Chem. 2009, 284, 10034–10045. [Google Scholar]
- Glenney, J.R., Jr.; Geisler, N.; Kaulfus, P.; Weber, K. Demonstration of at least two different actin-binding sites in villin, a calcium-regulated modulator of F-actin organization. J. Biol. Chem. 1981, 256, 8156–8161. [Google Scholar]
- Athman, R.; Louvard, D.; Robine, S. Villin enhances hepatocyte growth factor-induced actin cytoskeleton remodeling in epithelial cells. Mol. Biol. Cell 2003, 14, 4641–4653. [Google Scholar] [CrossRef]
- Bretscher, A.; Weber, K. Villin is a major protein of the microvillus cytoskeleton which binds both G and F actin in a calcium-dependent manner. Cell 1980, 20, 839–847. [Google Scholar] [CrossRef]
- Glenney, J.R., Jr.; Kaulfus, P.; Matsudaira, P.; Weber, K. F-actin binding and bundling properties of fimbrin, a major cytoskeletal protein of microvillus core filaments. J. Biol. Chem. 1981, 256, 9283–9288. [Google Scholar]
- Glenney, J.R., Jr.; Kaulfus, P.; Weber, K. F actin assembly modulated by villin: Ca+-dependent nucleation and capping of the barbed end. Cell 1981, 24, 471–480. [Google Scholar] [CrossRef]
- Ubelmann, F.; Chamaillard, M.; El-Marjou, F.; Simon, A.; Netter, J.; Vignjevic, D.; Nichols, B.L.; Quezada-Calvillo, R.; Grandjean, T.; Louvard, D.; et al. Enterocyte loss of polarity and gut wound healing rely upon the F-actin-severing function of villin. Proc. Natl. Acad. Sci. USA 2013, 110, E1380–E1389. [Google Scholar] [CrossRef]
- Ferrary, E.; Cohen-Tannoudji, M.; Pehau-Arnaudet, G.; Lapillonne, A.; Athman, R.; Ruiz, T.; Boulouha, L.; El Marjou, F.; Doye, A.; Fontaine, J.J.; et al. In vivo, villin is required for ca2+-dependent F-actin disruption in intestinal brush borders. J. Cell Biol. 1999, 146, 819–830. [Google Scholar] [CrossRef]
- Wang, Y.; Srinivasan, K.; Siddiqui, M.R.; George, S.P.; Tomar, A.; Khurana, S. A novel role for villin in intestinal epithelial cell survival and homeostasis. J. Biol. Chem. 2008, 283, 9454–9464. [Google Scholar] [CrossRef]
- Kersting, S.; Bruewer, M.; Schuermann, G.; Klotz, A.; Utech, M.; Hansmerten, M.; Krieglstein, C.F.; Senninger, N.; Schulzke, J.D.; Naim, H.Y.; et al. Antigen transport and cytoskeletal characteristics of a distinct enterocyte population in inflammatory bowel diseases. Am. J. Pathol. 2004, 165, 425–437. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Elson, C.O.; Tennyson, G.S.; Beagley, K.W. Kinetics of cytokine expression during healing of acute colitis in mice. Am. J. Pathol. 1996, 271, G130–G136. [Google Scholar]
- Perse, M.; Cerar, A. Dextran sodium sulphate colitis mouse model: Traps and tricks. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Dignass, A.U.; Podolsky, D.K. Cytokine modulation of intestinal epithelial cell restitution: Central role of transforming growth factor β. Gastroenterology 1993, 105, 1323–1332. [Google Scholar] [CrossRef]
- Playford, R.J.; Marchbank, T.; Chinery, R.; Evison, R.; Pignatelli, M.; Boulton, R.A.; Thim, L.; Hanby, A.M. Human spasmolytic polypeptide is a cytoprotective agent that stimulates cell migration. Gastroenterology 1995, 108, 108–116. [Google Scholar] [CrossRef]
- Paclik, D.; Lohse, K.; Wiedenmann, B.; Dignass, A.U.; Sturm, A. Galectin-2 and -4, but not galectin-1, promote intestinal epithelial wound healing in vitro through a TGF-β-independent mechanism. Inflamm. Bowel Dis. 2008, 14, 1366–1372. [Google Scholar] [CrossRef]
- Cao, Z.; Said, N.; Amin, S.; Wu, H.K.; Bruce, A.; Garate, M.; Hsu, D.K.; Kuwabara, I.; Liu, F.T.; Panjwani, N. Galectins-3 and -7, but not galectin-1, play a role in re-epithelialization of wounds. J. Biol. Chem. 2002, 277, 42299–42305. [Google Scholar]
- Puthenedam, M.; Wu, F.; Shetye, A.; Michaels, A.; Rhee, K.J.; Kwon, J.H. Matrilysin-1 (MMP7) cleaves galectin-3 and inhibits wound healing in intestinal epithelial cells. Inflamm. Bowel Dis. 2011, 17, 260–267. [Google Scholar] [CrossRef]
- Wong, W.M.; Poulsom, R.; Wright, N.A. Trefoil peptides. Gut 1999, 44, 890–895. [Google Scholar] [CrossRef]
- Taupin, D.; Podolsky, D.K. Trefoil factors: Initiators of mucosal healing. Nat. Rev. Mol. Cell Biol. 2003, 4, 721–732. [Google Scholar] [CrossRef]
- Mashimo, H.; Wu, D.C.; Podolsky, D.K.; Fishman, M.C. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 1996, 274, 262–265. [Google Scholar] [CrossRef]
- Playford, R.J.; Marchbank, T.; Goodlad, R.A.; Chinery, R.A.; Poulsom, R.; Hanby, A.M. Transgenic mice that overexpress the human trefoil peptide PS2 have an increased resistance to intestinal damage. Proc. Natl. Acad. Sci. USA 1996, 93, 2137–2142. [Google Scholar] [CrossRef]
- Tran, C.P.; Cook, G.A.; Yeomans, N.D.; Thim, L.; Giraud, A.S. Trefoil peptide TFF2 (spasmolytic polypeptide) potently accelerates healing and reduces inflammation in a rat model of colitis. Gut 1999, 44, 636–642. [Google Scholar] [CrossRef]
- Poulsen, S.S.; Kissow, H.; Hare, K.; Hartmann, B.; Thim, L. Luminal and parenteral TFF2 and TFF3 dimer and monomer in two models of experimental colitis in the rat. Regul. Pept. 2005, 126, 163–171. [Google Scholar] [CrossRef]
- Soriano-Izquierdo, A.; Gironella, M.; Massaguer, A.; May, F.E.; Salas, A.; Sans, M.; Poulsom, R.; Thim, L.; Pique, J.M.; Panes, J. Trefoil peptide TFF2 treatment reduces VCAM-1 expression and leukocyte recruitment in experimental intestinal inflammation. J. Leukoc. Biol. 2004, 75, 214–223. [Google Scholar]
- Taupin, D.; Wu, D.C.; Jeon, W.K.; Devaney, K.; Wang, T.C.; Podolsky, D.K. The trefoil gene family are coordinately expressed immediate-early genes: EGF receptor- and map kinase-dependent interregulation. J. Clin. Investig. 1999, 103, R31–R38. [Google Scholar] [CrossRef]
- Taupin, D.R.; Kinoshita, K.; Podolsky, D.K. Intestinal trefoil factor confers colonic epithelial resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 799–804. [Google Scholar] [CrossRef]
- Kinoshita, K.; Taupin, D.R.; Itoh, H.; Podolsky, D.K. Distinct pathways of cell migration and antiapoptotic response to epithelial injury: Structure-function analysis of human intestinal trefoil factor. Mol. Cell. Biol. 2000, 20, 4680–4690. [Google Scholar] [CrossRef]
- Loncar, M.B.; Al-azzeh, E.D.; Sommer, P.S.; Marinovic, M.; Schmehl, K.; Kruschewski, M.; Blin, N.; Stohwasser, R.; Gott, P.; Kayademir, T. Tumour necrosis factor α and nuclear factor κb inhibit transcription of human TFF3 encoding a gastrointestinal healing peptide. Gut 2003, 52, 1297–1303. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-κb: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial nemo links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef]
- Egan, L.J.; Eckmann, L.; Greten, F.R.; Chae, S.; Li, Z.W.; Myhre, G.M.; Robine, S.; Karin, M.; Kagnoff, M.F. Iκb-kinaseβ-dependent NF-κb activation provides radioprotection to the intestinal epithelium. Proc. Natl. Acad. Sci. USA 2004, 101, 2452–2457. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-κb regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Spehlmann, M.E.; Eckmann, L. Nuclear factor-κ b in intestinal protection and destruction. Curr. Opin. Gastroenterol. 2009, 25, 92–99. [Google Scholar] [CrossRef]
- Dignass, A.U.; Lynch-Devaney, K.; Podolsky, D.K. Hepatocyte growth factor/scatter factor modulates intestinal epithelial cell proliferation and migration. Biochem. Biophys. Res. Commun. 1994, 202, 701–709. [Google Scholar] [CrossRef]
- Dignass, A.U.; Tsunekawa, S.; Podolsky, D.K. Fibroblast growth factors modulate intestinal epithelial cell growth and migration. Gastroenterology 1994, 106, 1254–1262. [Google Scholar]
- Housley, R.M.; Morris, C.F.; Boyle, W.; Ring, B.; Biltz, R.; Tarpley, J.E.; Aukerman, S.L.; Devine, P.L.; Whitehead, R.H.; Pierce, G.F. Keratinocyte growth factor induces proliferation of hepatocytes and epithelial cells throughout the rat gastrointestinal tract. J. Clin. Investig. 1994, 94, 1764–1777. [Google Scholar] [CrossRef]
- Ohneda, K.; Ulshen, M.H.; Fuller, C.R.; D’Ercole, A.J.; Lund, P.K. Enhanced growth of small bowel in transgenic mice expressing human insulin-like growth factor I. Gastroenterology 1997, 112, 444–454. [Google Scholar] [CrossRef]
- Dignass, A.U.; Podolsky, D.K. Interleukin 2 modulates intestinal epithelial cell function in vitro. Exp. Cell Res. 1996, 225, 422–429. [Google Scholar] [CrossRef]
- Ciacci, C.; Lind, S.E.; Podolsky, D.K. Transforming growth factor β regulation of migration in wounded rat intestinal epithelial monolayers. Gastroenterology 1993, 105, 93–101. [Google Scholar]
- Liu, F.; Wu, H.Y.; Wesselschmidt, R.; Kornaga, T.; Link, D.C. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity 1996, 5, 491–501. [Google Scholar] [CrossRef]
- Ramsay, R.G.; Micallef, S.J.; Williams, B.; Lightowler, S.; Vincan, E.; Heath, J.K.; Mantamadiotis, T.; Bertoncello, I. Colony-stimulating factor-1 promotes clonogenic growth of normal murine colonic crypt epithelial cells in vitro. J. Interferon Cytokine Res. 2004, 24, 416–427. [Google Scholar] [CrossRef]
- Egger, B.; Procaccino, F.; Sarosi, I.; Tolmos, J.; Buchler, M.W.; Eysselein, V.E. Keratinocyte growth factor ameliorates dextran sodium sulfate colitis in mice. Dig. Dis. Sci. 1999, 44, 836–844. [Google Scholar] [CrossRef]
- Greenwood-Van Meerveld, B.; Venkova, K.; Connolly, K. Efficacy of repifermin (keratinocyte growth factor-2) against abnormalities in gastrointestinal mucosal transport in a murine model of colitis. J. Pharm. Pharmacol. 2003, 55, 67–75. [Google Scholar] [CrossRef]
- Scaldaferri, F.; Vetrano, S.; Sans, M.; Arena, V.; Straface, G.; Stigliano, E.; Repici, A.; Sturm, A.; Malesci, A.; Panes, J.; et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology 2009, 136, 585–595. [Google Scholar] [CrossRef]
- Nair, D.G.; Miller, K.G.; Lourenssen, S.R.; Blennerhassett, M.G. Inflammatory cytokines promote growth of intestinal smooth muscle cells by induced expression of PDGF-Rβ. J. Cell. Mol. Med. 2014, 18, 444–454. [Google Scholar] [CrossRef]
- Sainathan, S.K.; Hanna, E.M.; Gong, Q.; Bishnupuri, K.S.; Luo, Q.; Colonna, M.; White, F.V.; Croze, E.; Houchen, C.; Anant, S.; et al. Granulocyte macrophage colony-stimulating factor ameliorates dss-induced experimental colitis. Inflamm. Bowel Dis. 2008, 14, 88–99. [Google Scholar] [CrossRef]
- Hoffmann, P.; Reinshagen, M.; Zeeh, J.M.; Lakshmanan, J.; Wu, V.S.; Goebell, H.; Gerken, G.; Eysselein, V.E. Increased expression of epidermal growth factor-receptor in an experimental model of colitis in rats. Scand. J. Gastroenterol. 2000, 35, 1174–1180. [Google Scholar] [CrossRef]
- Oikonomou, K.A.; Kapsoritakis, A.N.; Kapsoritaki, A.I.; Manolakis, A.C.; Tsiopoulos, F.D.; Germenis, A.E.; Potamianos, S.P. Downregulation of serum epidermal growth factor in patients with inflammatory bowel disease. Is there a link with mucosal damage? Growth F. 2010, 28, 461–466. [Google Scholar] [CrossRef]
- Hormi, K.; Cadiot, G.; Kermorgant, S.; Dessirier, V.; le Romancer, M.; Lewin, M.J.; Mignon, M.; Lehy, T. Transforming growth factor-α and epidermal growth factor receptor in colonic mucosa in active and inactive inflammatory bowel disease. Growth Factors 2000, 18, 79–91. [Google Scholar] [CrossRef]
- Frey, M.R.; Golovin, A.; Polk, D.B. Epidermal growth factor-stimulated intestinal epithelial cell migration requires SRC family kinase-dependent p38 MAPK signaling. J. Biol. Chem. 2004, 279, 44513–44521. [Google Scholar] [CrossRef]
- Frey, M.R.; Dise, R.S.; Edelblum, K.L.; Polk, D.B. P38 kinase regulates epidermal growth factor receptor downregulation and cellular migration. EMBO J. 2006, 25, 5683–5692. [Google Scholar] [CrossRef]
- Yamaoka, T.; Yan, F.; Cao, H.; Hobbs, S.S.; Dise, R.S.; Tong, W.; Polk, D.B. Transactivation of EGF receptor and ERBB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11772–11777. [Google Scholar]
- Hoffmann, P.; Zeeh, J.M.; Lakshmanan, J.; Wu, V.S.; Procaccino, F.; Reinshagen, M.; McRoberts, J.A.; Eysselein, V.E. Increased expression of transforming growth factor α precursors in acute experimental colitis in rats. Gut 1997, 41, 195–202. [Google Scholar] [CrossRef]
- Procaccino, F.; Reinshagen, M.; Hoffmann, P.; Zeeh, J.M.; Lakshmanan, J.; McRoberts, J.A.; Patel, A.; French, S.; Eysselein, V.E. Protective effect of epidermal growth factor in an experimental model of colitis in rats. Gastroenterology 1994, 107, 12–17. [Google Scholar]
- Nishimura, T.; Andoh, A.; Nishida, A.; Shioya, M.; Koizumi, Y.; Tsujikawa, T.; Fujiyama, Y. Fr167653, a p38 mitogen-activated protein kinase inhibitor, aggravates experimental colitis in mice. World J. Gastroenterol. 2008, 14, 5851–5856. [Google Scholar] [CrossRef]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Karlsson, S. Transforming growth factor-β 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am. J. Pathol. 1993, 143, 3–9. [Google Scholar]
- Fiocchi, C. TGF-β/smad signaling defects in inflammatory bowel disease: Mechanisms and possible novel therapies for chronic inflammation. J. Clin. Investig. 2001, 108, 523–526. [Google Scholar] [CrossRef]
- Monteleone, G.; Pallone, F.; MacDonald, T.T. SMAD7 in TGF-β-mediated negative regulation of gut inflammation. Trends Immun. 2004, 25, 513–517. [Google Scholar] [CrossRef]
- Feng, X.H.; Derynck, R. Specificity and versatility in TGF-β signaling through SMADs. Ann. Rev. Cell Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef]
- Beck, P.L.; Rosenberg, I.M.; Xavier, R.J.; Koh, T.; Wong, J.F.; Podolsky, D.K. Transforming growth factor-β mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am. J. Pathol. 2003, 162, 597–608. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RHOA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Kim, S.I.; Kwak, J.H.; Na, H.J.; Kim, J.K.; Ding, Y.; Choi, M.E. Transforming growth factor-β (TGF-β1) activates TAK1 via TAB1-mediated autophosphorylation, independent of TGF-β receptor kinase activity in mesangial cells. J. Biol. Chem. 2009, 284, 22285–22296. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kajino-Sakamoto, R.; Omori, E.; Jobin, C.; Ninomiya-Tsuji, J. Intestinal epithelial-derived TAK1 signaling is essential for cytoprotection against chemical-induced colitis. PLoS One 2009, 4. [Google Scholar] [CrossRef]
- Kajino-Sakamoto, R.; Inagaki, M.; Lippert, E.; Akira, S.; Robine, S.; Matsumoto, K.; Jobin, C.; Ninomiya-Tsuji, J. Enterocyte-derived TAK1 signaling prevents epithelium apoptosis and the development of ileitis and colitis. J. Immunol. 2008, 181, 1143–1152. [Google Scholar]
- Stappenbeck, T.S.; Miyoshi, H. The role of stromal stem cells in tissue regeneration and wound repair. Science 2009, 324, 1666–1669. [Google Scholar] [CrossRef]
- Barrett, T.A. Developmental biology. Intestinal wound healing requires a WNT balancing act. Science 2012, 338, 51–52. [Google Scholar] [CrossRef]
- Miyoshi, H.; Ajima, R.; Luo, C.T.; Yamaguchi, T.P.; Stappenbeck, T.S. WNT5A potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science 2012, 338, 108–113. [Google Scholar] [CrossRef]
- Moses, H.L.; Yang, E.Y.; Pietenpol, J.A. TGF-β stimulation and inhibition of cell proliferation: New mechanistic insights. Cell 1990, 63, 245–247. [Google Scholar] [CrossRef]
- Tessner, T.G.; Cohn, S.M.; Schloemann, S.; Stenson, W.F. Prostaglandins prevent decreased epithelial cell proliferation associated with dextran sodium sulfate injury in mice. Gastroenterology 1998, 115, 874–882. [Google Scholar] [CrossRef]
- Powell, D.W.; Saada, J.I. Mesenchymal stem cells and prostaglandins may be critical for intestinal wound repair. Gastroenterology 2012, 143, 19–22. [Google Scholar] [CrossRef]
- Newberry, R.D.; Stenson, W.F.; Lorenz, R.G. Cyclooxygenase-2-dependent arachidonic acid metabolites are essential modulators of the intestinal immune response to dietary antigen. Nat. Med. 1999, 5, 900–906. [Google Scholar]
- Cohn, S.M.; Schloemann, S.; Tessner, T.; Seibert, K.; Stenson, W.F. Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J. Clin. Investig. 1997, 99, 1367–1379. [Google Scholar] [CrossRef]
- Brown, S.L.; Riehl, T.E.; Walker, M.R.; Geske, M.J.; Doherty, J.M.; Stenson, W.F.; Stappenbeck, T.S. Myd88-dependent positioning of PTGS2-expressing stromal cells maintains colonic epithelial proliferation during injury. J. Clin. Investig. 2007, 117, 258–269. [Google Scholar] [CrossRef]
- Morteau, O.; Morham, S.G.; Sellon, R.; Dieleman, L.A.; Langenbach, R.; Smithies, O.; Sartor, R.B. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J. Clin. Investig. 2000, 105, 469–478. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Hao, L.; Medzhitov, R. Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity 2006, 25, 319–329. [Google Scholar] [CrossRef]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar]
- Nell, S.; Suerbaum, S.; Josenhans, C. The impact of the microbiota on the pathogenesis of IBD: Lessons from mouse infection models. Nat. Rev. Microbiol. 2010, 8, 564–577. [Google Scholar] [CrossRef]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef]
- Abreu, M.T.; Vora, P.; Faure, E.; Thomas, L.S.; Arnold, E.T.; Arditi, M. Decreased expression of toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J. Immunol. 2001, 167, 1609–1616. [Google Scholar] [CrossRef]
- Furrie, E.; Macfarlane, S.; Thomson, G.; Macfarlane, G.T.; Microbiology; Gut Biology, G.; Tayside, T.; Tumour, B. Toll-like receptors-2, -3 and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology 2005, 115, 565–574. [Google Scholar] [CrossRef]
- Melmed, G.; Thomas, L.S.; Lee, N.; Tesfay, S.Y.; Lukasek, K.; Michelsen, K.S.; Zhou, Y.; Hu, B.; Arditi, M.; Abreu, M.T. Human intestinal epithelial cells are broadly unresponsive to toll-like receptor 2-dependent bacterial ligands: Implications for host-microbial interactions in the gut. J. Immunol. 2003, 170, 1406–1415. [Google Scholar] [CrossRef]
- Cario, E.; Brown, D.; McKee, M.; Lynch-Devaney, K.; Gerken, G.; Podolsky, D.K. Commensal-associated molecular patterns induce selective toll-like receptor-trafficking from apical membrane to cytoplasmic compartments in polarized intestinal epithelium. Am. J. Pathol. 2002, 160, 165–173. [Google Scholar]
- Ortega-Cava, C.F.; Ishihara, S.; Rumi, M.A.; Kawashima, K.; Ishimura, N.; Kazumori, H.; Udagawa, J.; Kadowaki, Y.; Kinoshita, Y. Strategic compartmentalization of toll-like receptor 4 in the mouse gut. J. Immunol. 2003, 170, 3977–3985. [Google Scholar] [CrossRef]
- Hornef, M.W.; Frisan, T.; Vandewalle, A.; Normark, S.; Richter-Dahlfors, A. Toll-like receptor 4 resides in the golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J. Exp. Med. 2002, 195, 559–570. [Google Scholar] [CrossRef]
- Hornef, M.W.; Normark, B.H.; Vandewalle, A.; Normark, S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J. Exp. Med. 2003, 198, 1225–1235. [Google Scholar] [CrossRef]
- Cario, E. Toll-like receptors in inflammatory bowel diseases: A decade later. Inflamm. Bowel Dis. 2010, 16, 1583–1597. [Google Scholar] [CrossRef]
- Gibson, D.L.; Ma, C.; Bergstrom, K.S.; Huang, J.T.; Man, C.; Vallance, B.A. MYD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during citrobacter rodentium-induced colitis. Cell. Microbiol. 2008, 10, 618–631. [Google Scholar] [CrossRef]
- Fukata, M.; Breglio, K.; Chen, A.; Vamadevan, A.S.; Goo, T.; Hsu, D.; Conduah, D.; Xu, R.; Abreu, M.T. The myeloid differentiation factor 88 (MYD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J. Immunol. 2008, 180, 1886–1894. [Google Scholar] [CrossRef]
- Tomita, T.; Kanai, T.; Fujii, T.; Nemoto, Y.; Okamoto, R.; Tsuchiya, K.; Totsuka, T.; Sakamoto, N.; Akira, S.; Watanabe, M. MYD88-dependent pathway in T cells directly modulates the expansion of colitogenic CD4+ T cells in chronic colitis. J. Immunol. 2008, 180, 5291–5299. [Google Scholar] [CrossRef]
- Asquith, M.J.; Boulard, O.; Powrie, F.; Maloy, K.J. Pathogenic and protective roles of MYD88 in leukocytes and epithelial cells in mouse models of inflammatory bowel disease. Gastroenterology 2010, 139, 519–529. [Google Scholar] [CrossRef]
- Hapfelmeier, S.; Stecher, B.; Barthel, M.; Kremer, M.; Muller, A.J.; Heikenwalder, M.; Stallmach, T.; Hensel, M.; Pfeffer, K.; Akira, S.; et al. The salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow salmonella serovar typhimurium to trigger colitis via MYD88-dependent and MYD88-independent mechanisms. J. Immunol. 2005, 174, 1675–1685. [Google Scholar] [CrossRef]
- Fukata, M.; Michelsen, K.S.; Eri, R.; Thomas, L.S.; Hu, B.; Lukasek, K.; Nast, C.C.; Lechago, J.; Xu, R.; Naiki, Y.; et al. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. 2005, 288, G1055–G1065. [Google Scholar]
- Vijay-Kumar, M.; Sanders, C.J.; Taylor, R.T.; Kumar, A.; Aitken, J.D.; Sitaraman, S.V.; Neish, A.S.; Uematsu, S.; Akira, S.; Williams, I.R.; et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Investig. 2007, 117, 3909–3921. [Google Scholar]
- Lee, J.; Mo, J.H.; Katakura, K.; Alkalay, I.; Rucker, A.N.; Liu, Y.T.; Lee, H.K.; Shen, C.; Cojocaru, G.; Shenouda, S.; et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat. Cell Biol. 2006, 8, 1327–1336. [Google Scholar] [CrossRef]
- Ey, B.; Eyking, A.; Klepak, M.; Salzman, N.H.; Gothert, J.R.; Runzi, M.; Schmid, K.W.; Gerken, G.; Podolsky, D.K.; Cario, E. Loss of TLR2 worsens spontaneous colitis in MDR1A deficiency through commensally induced pyroptosis. J. Immunol. 2013, 190, 5676–5688. [Google Scholar]
- Rachmilewitz, D.; Katakura, K.; Karmeli, F.; Hayashi, T.; Reinus, C.; Rudensky, B.; Akira, S.; Takeda, K.; Lee, J.; Takabayashi, K.; et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology 2004, 126, 520–528. [Google Scholar] [CrossRef]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology 2004, 127, 224–238. [Google Scholar] [CrossRef]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 2007, 132, 1359–1374. [Google Scholar] [CrossRef]
- Fukata, M.; Chen, A.; Klepper, A.; Krishnareddy, S.; Vamadevan, A.S.; Thomas, L.S.; Xu, R.; Inoue, H.; Arditi, M.; Dannenberg, A.J.; et al. Cox-2 is regulated by toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology 2006, 131, 862–877. [Google Scholar] [CrossRef]
- Zheng, L.; Riehl, T.E.; Stenson, W.F. Regulation of colonic epithelial repair in mice by toll-like receptors and hyaluronic acid. Gastroenterology 2009, 137, 2041–2051. [Google Scholar] [CrossRef]
- Podolsky, D.K.; Gerken, G.; Eyking, A.; Cario, E. Colitis-associated variant of TLR2 causes impaired mucosal repair because of TFF3 deficiency. Gastroenterology 2009, 137, 209–220. [Google Scholar] [CrossRef]
- Fukata, M.; Abreu, M.T. TLR4 signalling in the intestine in health and disease. Biochem. Soc. Transact. 2007, 35, 1473–1478. [Google Scholar] [CrossRef]
- Brandl, K.; Sun, L.; Neppl, C.; Siggs, O.M.; le Gall, S.M.; Tomisato, W.; Li, X.; Du, X.; Maennel, D.N.; Blobel, C.P.; et al. MYD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 19967–19972. [Google Scholar] [CrossRef]
- Rose, W.A., 2nd; Sakamoto, K.; Leifer, C.A. TLR9 is important for protection against intestinal damage and for intestinal repair. Sci. Rep. 2012, 2, 574. [Google Scholar]
- Lin, N.; Xu, L.F.; Sun, M. The protective effect of trefoil factor 3 on the intestinal tight junction barrier is mediated by toll-like receptor 2 via a PI3K/AKT dependent mechanism. Biochem. Biophys. Res. Commun. 2013, 440, 143–149. [Google Scholar] [CrossRef]
- Pull, S.L.; Doherty, J.M.; Mills, J.C.; Gordon, J.I.; Stappenbeck, T.S. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc. Natl. Acad. Sci. USA 2005, 102, 99–104. [Google Scholar]
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef]
- Moore, C.B.; Bergstralh, D.T.; Duncan, J.A.; Lei, Y.; Morrison, T.E.; Zimmermann, A.G.; Accavitti-Loper, M.A.; Madden, V.J.; Sun, L.; Ye, Z.; et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008, 451, 573–577. [Google Scholar] [CrossRef]
- Tattoli, I.; Carneiro, L.A.; Jehanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κb and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008, 9, 293–300. [Google Scholar] [CrossRef]
- Meylan, E.; Tschopp, J. NLRX1: Friend or foe? EMBO Rep. 2008, 9, 243–245. [Google Scholar] [CrossRef]
- Chamaillard, M.; Hashimoto, M.; Horie, Y.; Masumoto, J.; Qiu, S.; Saab, L.; Ogura, Y.; Kawasaki, A.; Fukase, K.; Kusumoto, S.; et al. An essential role for NOD2 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 2003, 4, 702–707. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.; Antignac, A.; Jehanno, M.; Viala, J.; Tedin, K.; Taha, M.K.; Labigne, A.; Zahringer, U.; et al. NOD2 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef]
- McDonald, C.; Inohara, N.; Nunez, G. Peptidoglycan signaling in innate immunity and inflammatory disease. J. Biol. Chem. 2005, 280, 20177–20180. [Google Scholar] [CrossRef]
- Inohara, N.; Koseki, T.; Lin, J.; del Peso, L.; Lucas, P.C.; Chen, F.F.; Ogura, Y.; Nunez, G. An induced proximity model for NF-κb activation in the NOD2/rick and RIP signaling pathways. J. Biol. Chem. 2000, 275, 27823–27831. [Google Scholar]
- Park, J.H.; Kim, Y.G.; McDonald, C.; Kanneganti, T.D.; Hasegawa, M.; Body-Malapel, M.; Inohara, N.; Nunez, G. RICK/RIP2 mediates innate immune responses induced through NOD1 and NOD2 but not TLRS. J. Immunol. 2007, 178, 2380–2386. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Ellison, L.K.; Ramjeet, M.; Travassos, L.H.; Jones, N.L.; Girardin, S.E.; Philpott, D.J. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors NOD1 and NOD2 in an autophagy-independent manner. Immunity 2013, 39, 858–873. [Google Scholar] [CrossRef]
- Magalhaes, J.G.; Lee, J.; Geddes, K.; Rubino, S.; Philpott, D.J.; Girardin, S.E. Essential role of RIP2 in the modulation of innate and adaptive immunity triggered by NOD2 and NOD2 ligands. Eur. J. Immunol. 2011, 41, 1445–1455. [Google Scholar] [CrossRef]
- Krieg, A.; Correa, R.G.; Garrison, J.B.; le Negrate, G.; Welsh, K.; Huang, Z.; Knoefel, W.T.; Reed, J.C. XIAP mediates NOD signaling via interaction with RIP2. Proc. Natl. Acad. Sci. USA 2009, 106, 14524–14529. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Doiron, K.; Labbe, K.; Korneluk, R.G.; Barker, P.A.; Saleh, M. Cellular inhibitors of apoptosis CIAP1 and CIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 2009, 30, 789–801. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujimoto, Y.; Lucas, P.C.; Nakano, H.; Fukase, K.; Nunez, G.; Inohara, N. A critical role of RICK/RIP2 polyubiquitination in NOD-induced NF-κb activation. EMBO J. 2008, 27, 373–383. [Google Scholar] [CrossRef]
- Yang, Y.; Yin, C.; Pandey, A.; Abbott, D.; Sassetti, C.; Kelliher, M.A. NOD2 pathway activation by MDP or mycobacterium tuberculosis infection involves the stable polyubiquitination of RIP2. J. Biol. Chem. 2007, 282, 36223–36229. [Google Scholar]
- Tigno-Aranjuez, J.T.; Asara, J.M.; Abbott, D.W. Inhibition of RIP2’s tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 2010, 24, 2666–2677. [Google Scholar] [CrossRef]
- Lecine, P.; Esmiol, S.; Metais, J.Y.; Nicoletti, C.; Nourry, C.; McDonald, C.; Nunez, G.; Hugot, J.P.; Borg, J.P.; Ollendorff, V. The NOD2-RICK complex signals from the plasma membrane. J. Biol. Chem. 2007, 282, 15197–15207. [Google Scholar] [CrossRef]
- da Silva Correia, J.; Miranda, Y.; Leonard, N.; Hsu, J.; Ulevitch, R.J. Regulation of NOD2-mediated signaling pathways. Cell Death Differ. 2007, 14, 830–839. [Google Scholar] [CrossRef]
- Yeretssian, G.; Correa, R.G.; Doiron, K.; Fitzgerald, P.; Dillon, C.P.; Green, D.R.; Reed, J.C.; Saleh, M. Non-apoptotic role of BID in inflammation and innate immunity. Nature 2011, 474, 96–99. [Google Scholar] [CrossRef]
- Lipinski, S.; Grabe, N.; Jacobs, G.; Billmann-Born, S.; Till, A.; Hasler, R.; Aden, K.; Paulsen, M.; Arlt, A.; Kraemer, L.; et al. Rnai screening identifies mediators of NOD2 signaling: Implications for spatial specificity of mdp recognition. Proc. Natl. Acad. Sci. USA 2012, 109, 21426–21431. [Google Scholar] [CrossRef]
- Warner, N.; Burberry, A.; Franchi, L.; Kim, Y.G.; McDonald, C.; Sartor, M.A.; Nunez, G. A genome-wide sirna screen reveals positive and negative regulators of the NOD2 and NF-κb signaling pathways. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef]
- Askari, N.; Correa, R.G.; Zhai, D.; Reed, J.C. Expression, purification, and characterization of recombinant NOD2 (NLRC1): A NLR family member. J. Biotechnol. 2012, 157, 75–81. [Google Scholar] [CrossRef]
- Kim, Y.G.; Kamada, N.; Shaw, M.H.; Warner, N.; Chen, G.Y.; Franchi, L.; Nunez, G. The NOD2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 2011, 34, 769–780. [Google Scholar] [CrossRef]
- Correa, R.G.; Milutinovic, S.; Reed, J.C. Roles of NOD1 (NLRC1) and NOD2 (NLRC2) in innate immunity and inflammatory diseases. Biosci. Rep. 2012, 32, 597–608. [Google Scholar] [CrossRef]
- Ogura, Y.; Inohara, N.; Benito, A.; Chen, F.F.; Yamaoka, S.; Nunez, G. NOD2, a NOD2/APAF-1 family member that is restricted to monocytes and activates NF-κb. J. Biol. Chem. 2001, 276, 4812–4818. [Google Scholar]
- Barnich, N.; Aguirre, J.E.; Reinecker, H.C.; Xavier, R.; Podolsky, D.K. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-κb activation in muramyl dipeptide recognition. J. Cell Biol. 2005, 170, 21–26. [Google Scholar] [CrossRef]
- Ogura, Y.; Lala, S.; Xin, W.; Smith, E.; Dowds, T.A.; Chen, F.F.; Zimmermann, E.; Tretiakova, M.; Cho, J.H.; Hart, J.; et al. Expression of NOD2 in paneth cells: A possible link to crohn’s ileitis. Gut 2003, 52, 1591–1597. [Google Scholar] [CrossRef]
- Hisamatsu, T.; Suzuki, M.; Reinecker, H.C.; Nadeau, W.J.; McCormick, B.A.; Podolsky, D.K. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology 2003, 124, 993–1000. [Google Scholar]
- Leung, C.H.; Lam, W.; Ma, D.L.; Gullen, E.A.; Cheng, Y.C. Butyrate mediates nucleotide-binding and oligomerisation domain NOD2-dependent mucosal immune responses against peptidoglycan. Eur. J. Immunol. 2009, 39, 3529–3537. [Google Scholar] [CrossRef]
- Rosenstiel, P.; Fantini, M.; Brautigam, K.; Kuhbacher, T.; Waetzig, G.H.; Seegert, D.; Schreiber, S. TNF-α and IFN-γ regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 2003, 124, 1001–1009. [Google Scholar]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef]
- Hampe, J.; Cuthbert, A.; Croucher, P.J.; Mirza, M.M.; Mascheretti, S.; Fisher, S.; Frenzel, H.; King, K.; Hasselmeyer, A.; MacPherson, A.J.; et al. Association between insertion mutation in NOD2 gene and crohn’s disease in german and british populations. Lancet 2001, 357, 1925–1928. [Google Scholar] [CrossRef]
- Billmann-Born, S.; Till, A.; Arlt, A.; Lipinski, S.; Sina, C.; Latiano, A.; Annese, V.; Hasler, R.; Kerick, M.; Manke, T.; et al. Genome-wide expression profiling identifies an impairment of negative feedback signals in the crohn’s disease-associated NOD2 variant L1007fsinsC. J. Immunol. 2011, 186, 4027–4038. [Google Scholar] [CrossRef]
- McGovern, D.P.; Hysi, P.; Ahmad, T.; van Heel, D.A.; Moffatt, M.F.; Carey, A.; Cookson, W.O.; Jewell, D.P. Association between a complex insertion/deletion polymorphism in NOD2 (CARD4) and susceptibility to inflammatory bowel disease. Human Mol. Genet. 2005, 14, 1245–1250. [Google Scholar] [CrossRef]
- Tremelling, M.; Hancock, L.; Bredin, F.; Sharpstone, D.; Bingham, S.A.; Parkes, M. Complex insertion/deletion polymorphism in NOD2 (CARD4) is not associated with inflammatory bowel disease susceptibility in east anglia panel. Inflamm. Bowel Dis. 2006, 12, 967–971. [Google Scholar] [CrossRef]
- McDonald, C.; Chen, F.F.; Ollendorff, V.; Ogura, Y.; Marchetto, S.; Lecine, P.; Borg, J.P.; Nunez, G. A role for erbin in the regulation of NOD2-dependent NF-κb signaling. J. Biol. Chem. 2005, 280, 40301–40309. [Google Scholar] [CrossRef]
- Kufer, T.A.; Kremmer, E.; Banks, D.J.; Philpott, D.J. Role for erbin in bacterial activation of NOD2. Infect. Immun. 2006, 74, 3115–3124. [Google Scholar] [CrossRef]
- Aktories, K.; Schmidt, G.; Just, I. Rho GTPases as targets of bacterial protein toxins. Biol. Chem. 2000, 381, 421–426. [Google Scholar]
- Fukazawa, A.; Alonso, C.; Kurachi, K.; Gupta, S.; Lesser, C.F.; McCormick, B.A.; Reinecker, H.C. GEF-H1 mediated control of NOD2 dependent NF-κb activation by shigella effectors. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef]
- Keestra, A.M.; Winter, M.G.; Auburger, J.J.; Frassle, S.P.; Xavier, M.N.; Winter, S.E.; Kim, A.; Poon, V.; Ravesloot, M.M.; Waldenmaier, J.F.; et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD2. Nature 2013, 496, 233–237. [Google Scholar] [CrossRef]
- Boyer, L.; Magoc, L.; Dejardin, S.; Cappillino, M.; Paquette, N.; Hinault, C.; Charriere, G.M.; Ip, W.K.; Fracchia, S.; Hennessy, E.; et al. Pathogen-derived effectors trigger protective immunity via activation of the RAC2 enzyme and the IMD or RIP kinase signaling pathway. Immunity 2011, 35, 536–549. [Google Scholar] [CrossRef]
- Watanabe, T.; Asano, N.; Murray, P.J.; Ozato, K.; Tailor, P.; Fuss, I.J.; Kitani, A.; Strober, W. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J. Clin. Investig. 2008, 118, 545–559. [Google Scholar]
- Macho Fernandez, E.; Valenti, V.; Rockel, C.; Hermann, C.; Pot, B.; Boneca, I.G.; Grangette, C. Anti-inflammatory capacity of selected lactobacilli in experimental colitis is driven by NOD2-mediated recognition of a specific peptidoglycan-derived muropeptide. Gut 2011, 60, 1050–1059. [Google Scholar] [CrossRef]
- Chen, G.Y.; Shaw, M.H.; Redondo, G.; Nunez, G. The innate immune receptor NOD2 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008, 68, 10060–10067. [Google Scholar] [CrossRef]
- Watanabe, T.; Kitani, A.; Murray, P.J.; Wakatsuki, Y.; Fuss, I.J.; Strober, W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity 2006, 25, 473–485. [Google Scholar] [CrossRef]
- Barreau, F.; Meinzer, U.; Chareyre, F.; Berrebi, D.; Niwa-Kawakita, M.; Dussaillant, M.; Foligne, B.; Ollendorff, V.; Heyman, M.; Bonacorsi, S.; et al. CARD15/NOD2 is required for peyer’s patches homeostasis in mice. PLoS One 2007, 2. [Google Scholar] [CrossRef]
- Natividad, J.M.; Petit, V.; Huang, X.; de Palma, G.; Jury, J.; Sanz, Y.; Philpott, D.; Garcia Rodenas, C.L.; McCoy, K.D.; Verdu, E.F. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1−/−; Nod2−/− mice. Inflamm. Bowel Dis. 2012, 18, 1434–1446. [Google Scholar] [CrossRef]
- Couturier-Maillard, A.; Secher, T.; Rehman, A.; Normand, S.; de Arcangelis, A.; Haesler, R.; Huot, L.; Grandjean, T.; Bressenot, A.; Delanoye-Crespin, A.; et al. Nod2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Investig. 2013, 123, 700–711. [Google Scholar]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nunez, G.; Flavell, R.A. NOD2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005, 307, 731–734. [Google Scholar] [CrossRef]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef]
- Petnicki-Ocwieja, T.; Hrncir, T.; Liu, Y.J.; Biswas, A.; Hudcovic, T.; Tlaskalova-Hogenova, H.; Kobayashi, K.S. NOD2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 15813–15818. [Google Scholar] [CrossRef]
- Bouskra, D.; Brezillon, C.; Berard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD2 regulates intestinal homeostasis. Nature 2008, 456, 507–510. [Google Scholar] [CrossRef]
- Geddes, K.; Rubino, S.J.; Magalhaes, J.G.; Streutker, C.; le Bourhis, L.; Cho, J.H.; Robertson, S.J.; Kim, C.J.; Kaul, R.; Philpott, D.J.; et al. Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat. Med. 2011, 17, 837–844. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The pyrin-card protein asc is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1β-processing inflammasome with increased activity in muckle-wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and IPAF. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Chen, G.Y. Role of NLRP6 and NLRP12 in the maintenance of intestinal homeostasis. Eur. J. Immunol. 2014, 44, 321–327. [Google Scholar] [CrossRef]
- Tamura, K.; Fukuda, Y.; Sashio, H.; Takeda, N.; Bamba, H.; Kosaka, T.; Fukui, S.; Sawada, K.; Tamura, K.; Satomi, M.; et al. IL18 polymorphism is associated with an increased risk of crohn’s disease. J. Gastroenterol. 2002, 37, 111–116. [Google Scholar] [CrossRef]
- Zhernakova, A.; Festen, E.M.; Franke, L.; Trynka, G.; van Diemen, C.C.; Monsuur, A.J.; Bevova, M.; Nijmeijer, R.M.; van Slot, R.; Heijmans, R.; et al. Genetic analysis of innate immunity in crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet. 2008, 82, 1202–1210. [Google Scholar] [CrossRef]
- Bauer, C.; Loher, F.; Dauer, M.; Mayer, C.; Lehr, H.A.; Schonharting, M.; Hallwachs, R.; Endres, S.; Eigler, A. The ice inhibitor pralnacasan prevents DSS-induced colitis in C57BL/6 mice and suppresses IP-10 mRNA but not TNF-α mRNA expression. Dig. Dis. Sci. 2007, 52, 1642–1652. [Google Scholar] [CrossRef]
- Siegmund, B.; Fantuzzi, G.; Rieder, F.; Gamboni-Robertson, F.; Lehr, H.A.; Hartmann, G.; Dinarello, C.A.; Endres, S.; Eigler, A. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-γ and TNF-α production. Am. J. Physiol. Regul. 2001, 281, R1264–R1273. [Google Scholar]
- Ishikura, T.; Kanai, T.; Uraushihara, K.; Iiyama, R.; Makita, S.; Totsuka, T.; Yamazaki, M.; Sawada, T.; Nakamura, T.; Miyata, T.; et al. Interleukin-18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin-18 transgenic mice. J. Gastroenterol. Hepatol. 2003, 18, 960–969. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Yeretssian, G.; Doiron, K.; Bergstrom, K.S.; McIntire, C.R.; LeBlanc, P.M.; Meunier, C.; Turbide, C.; Gros, P.; Beauchemin, N.; et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 2010, 32, 367–378. [Google Scholar] [CrossRef]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.M.; Wang, E.; Ma, W.; Haines, D.; O’HUigin, C.; et al. MYD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Zaki, M.H.; Vogel, P.; Body-Malapel, M.; Lamkanfi, M.; Kanneganti, T.D. IL-18 production downstream of the NLRP3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 2010, 185, 4912–4920. [Google Scholar] [CrossRef]
- Hirota, S.A.; Ng, J.; Lueng, A.; Khajah, M.; Parhar, K.; Li, Y.; Lam, V.; Potentier, M.S.; Ng, K.; Bawa, M.; et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 2011, 17, 1359–1372. [Google Scholar] [CrossRef]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar]
- Liu, Z.; Zaki, M.H.; Vogel, P.; Gurung, P.; Finlay, B.B.; Deng, W.; Lamkanfi, M.; Kanneganti, T.D. Role of inflammasomes in host defense against citrobacter rodentium infection. J. Biol. Chem. 2012, 287, 16955–16964. [Google Scholar]
- Song-Zhao, G.X.; Srinivasan, N.; Pott, J.; Baban, D.; Frankel, G.; Maloy, K.J. NLRP3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol. 2013, 2013. [Google Scholar] [CrossRef]
- Nordlander, S.; Pott, J.; Maloy, K.J. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol. 2013, 2013. [Google Scholar] [CrossRef]
- Lebeis, S.L.; Powell, K.R.; Merlin, D.; Sherman, M.A.; Kalman, D. Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen citrobacter rodentium. Infect. Immun. 2009, 77, 604–614. [Google Scholar] [CrossRef]
- Takagi, H.; Kanai, T.; Okazawa, A.; Kishi, Y.; Sato, T.; Takaishi, H.; Inoue, N.; Ogata, H.; Iwao, Y.; Hoshino, K.; et al. Contrasting action of Il-12 and Il-18 in the development of dextran sodium sulphate colitis in mice. Scand. J. Gastroenterol. 2003, 38, 837–844. [Google Scholar] [CrossRef]
- Siegmund, B. Interleukin-18 in intestinal inflammation: Friend and foe? Immunity 2010, 32, 300–302. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Byrne, B.G.; Whitfield, N.N.; Madigan, C.A.; Fuse, E.T.; Tateda, K.; Swanson, M.S. Cytosolic recognition of flagellin by mouse macrophages restricts legionella pneumophila infection. J. Exp. Med. 2006, 203, 1093–1104. [Google Scholar] [CrossRef]
- Miao, E.A.; Mao, D.P.; Yudkovsky, N.; Bonneau, R.; Lorang, C.G.; Warren, S.E.; Leaf, I.A.; Aderem, A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2010, 107, 3076–3080. [Google Scholar] [CrossRef]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via IPAF. Nat. Immunol. 2006, 7, 569–575. [Google Scholar] [CrossRef]
- Carvalho, F.A.; Nalbantoglu, I.; Aitken, J.D.; Uchiyama, R.; Su, Y.; Doho, G.H.; Vijay-Kumar, M.; Gewirtz, A.T. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol. 2012, 5, 288–298. [Google Scholar]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef]
- Normand, S.; Delanoye-Crespin, A.; Bressenot, A.; Huot, L.; Grandjean, T.; Peyrin-Biroulet, L.; Lemoine, Y.; Hot, D.; Chamaillard, M. NOD-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl. Acad. Sci. USA 2011, 108, 9601–9606. [Google Scholar] [CrossRef]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Nunez, G. A functional role for nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar]
- Zaki, M.H.; Vogel, P.; Malireddi, R.K.; Body-Malapel, M.; Anand, P.K.; Bertin, J.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 2011, 20, 649–660. [Google Scholar] [CrossRef]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.M.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κb signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef]
- Wang, L.; Manji, G.A.; Grenier, J.M.; Al-Garawi, A.; Merriam, S.; Lora, J.M.; Geddes, B.J.; Briskin, M.; DiStefano, P.S.; Bertin, J. PYPAF7, a novel pyrin-containing APAF1-like protein that regulates activation of NF-κb and caspase-1-dependent cytokine processing. J. Biol. Chem. 2002, 277, 29874–29880. [Google Scholar] [CrossRef]
- Grenier, J.M.; Wang, L.; Manji, G.A.; Huang, W.J.; Al-Garawi, A.; Kelly, R.; Carlson, A.; Merriam, S.; Lora, J.M.; Briskin, M.; et al. Functional screening of five pypaf family members identifies PYPAF5 as a novel regulator of NF-κb and caspase-1. FEBS Lett. 2002, 530, 73–78. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Alnemri, E.S. Assembly, purification, and assay of the activity of the ASC pyroptosome. Method. Enzym. 2008, 442, 251–270. [Google Scholar] [CrossRef]
- Anand, P.K.; Malireddi, R.K.; Lukens, J.R.; Vogel, P.; Bertin, J.; Lamkanfi, M.; Kanneganti, T.D. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012, 488, 389–393. [Google Scholar] [CrossRef]
- Lich, J.D.; Williams, K.L.; Moore, C.B.; Arthur, J.C.; Davis, B.K.; Taxman, D.J.; Ting, J.P. Monarch-1 suppresses non-canonical NF-κb activation and p52-dependent chemokine expression in monocytes. J. Immunol. 2007, 178, 1256–1260. [Google Scholar] [CrossRef]
- Williams, K.L.; Lich, J.D.; Duncan, J.A.; Reed, W.; Rallabhandi, P.; Moore, C.; Kurtz, S.; Coffield, V.M.; Accavitti-Loper, M.A.; Su, L.; et al. The caterpiller protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor α-, and mycobacterium tuberculosis-induced pro-inflammatory signals. J. Biol. Chem. 2005, 280, 39914–39924. [Google Scholar] [CrossRef]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. Nlrp6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Parlato, M.; Yeretssian, G. NOD-Like Receptors in Intestinal Homeostasis and Epithelial Tissue Repair. Int. J. Mol. Sci. 2014, 15, 9594-9627. https://doi.org/10.3390/ijms15069594
Parlato M, Yeretssian G. NOD-Like Receptors in Intestinal Homeostasis and Epithelial Tissue Repair. International Journal of Molecular Sciences. 2014; 15(6):9594-9627. https://doi.org/10.3390/ijms15069594
Chicago/Turabian StyleParlato, Marianna, and Garabet Yeretssian. 2014. "NOD-Like Receptors in Intestinal Homeostasis and Epithelial Tissue Repair" International Journal of Molecular Sciences 15, no. 6: 9594-9627. https://doi.org/10.3390/ijms15069594