Genetic Diversity and Population Structure of the Critically Endangered Yangtze Finless Porpoise (Neophocaena asiaeorientalis asiaeorientalis) as Revealed by Mitochondrial and Microsatellite DNA


Abstract
:1. Introduction

2. Results
2.1. Genetic Diversity of the Yangtze Finless Porpoise

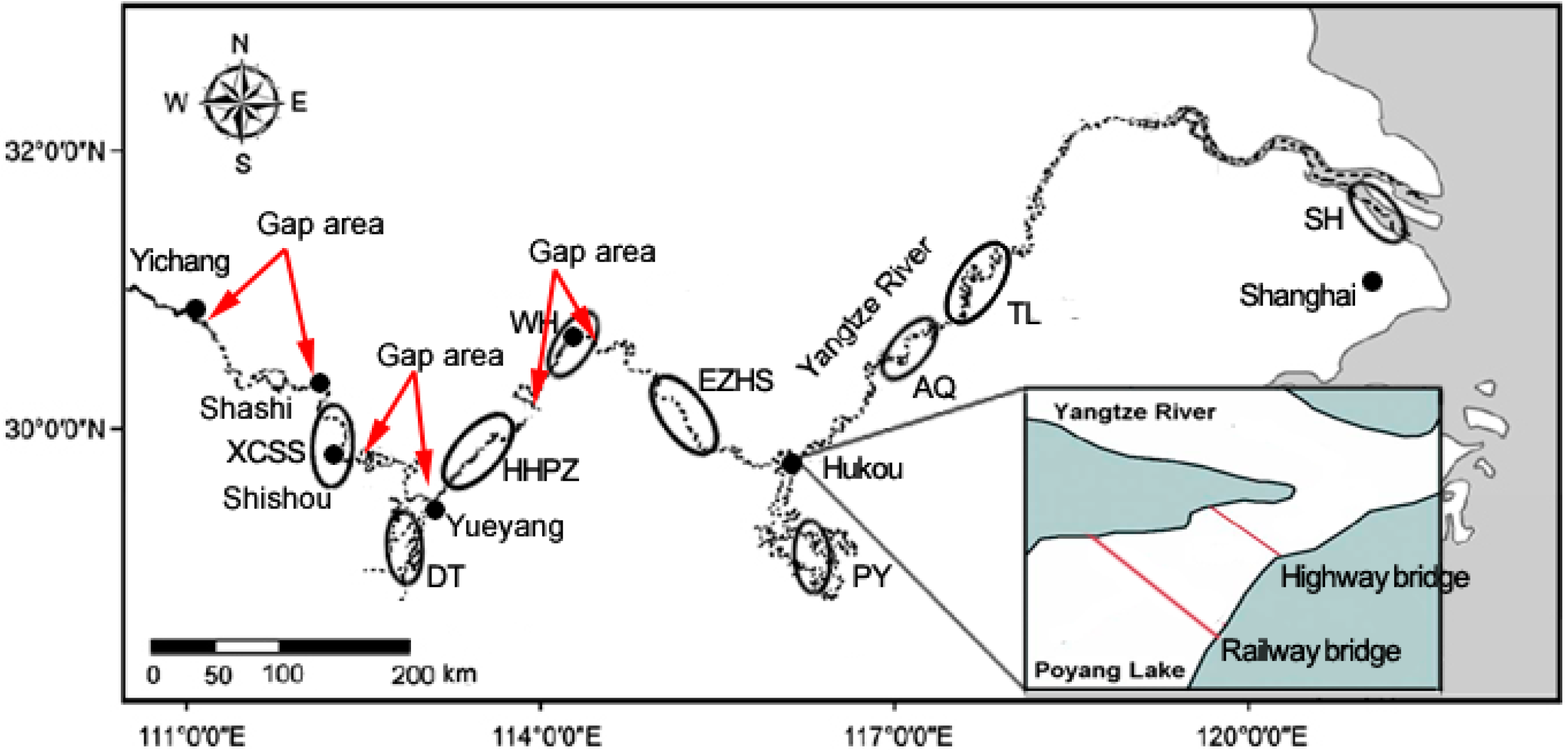{kind=link}
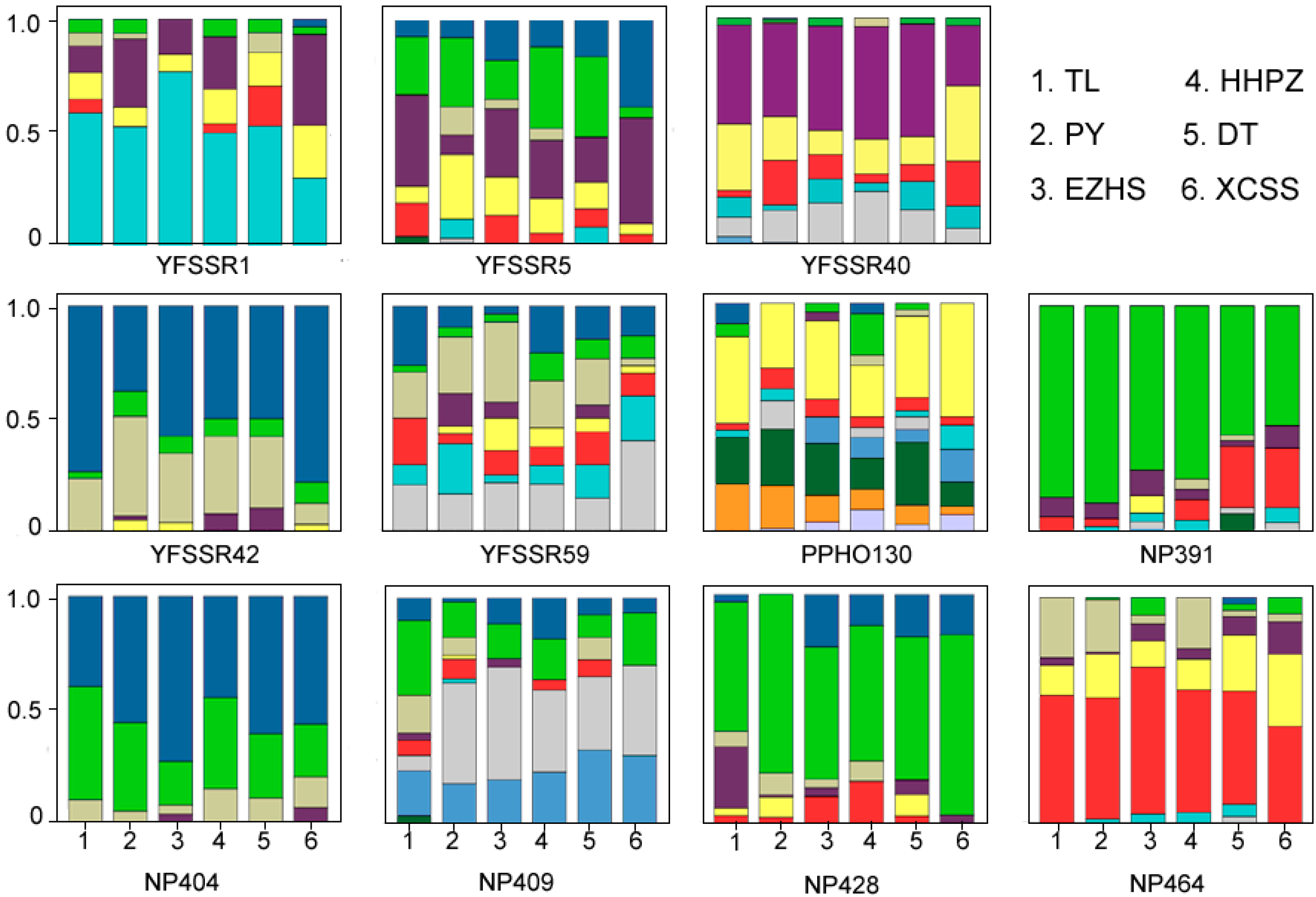{kind=link}
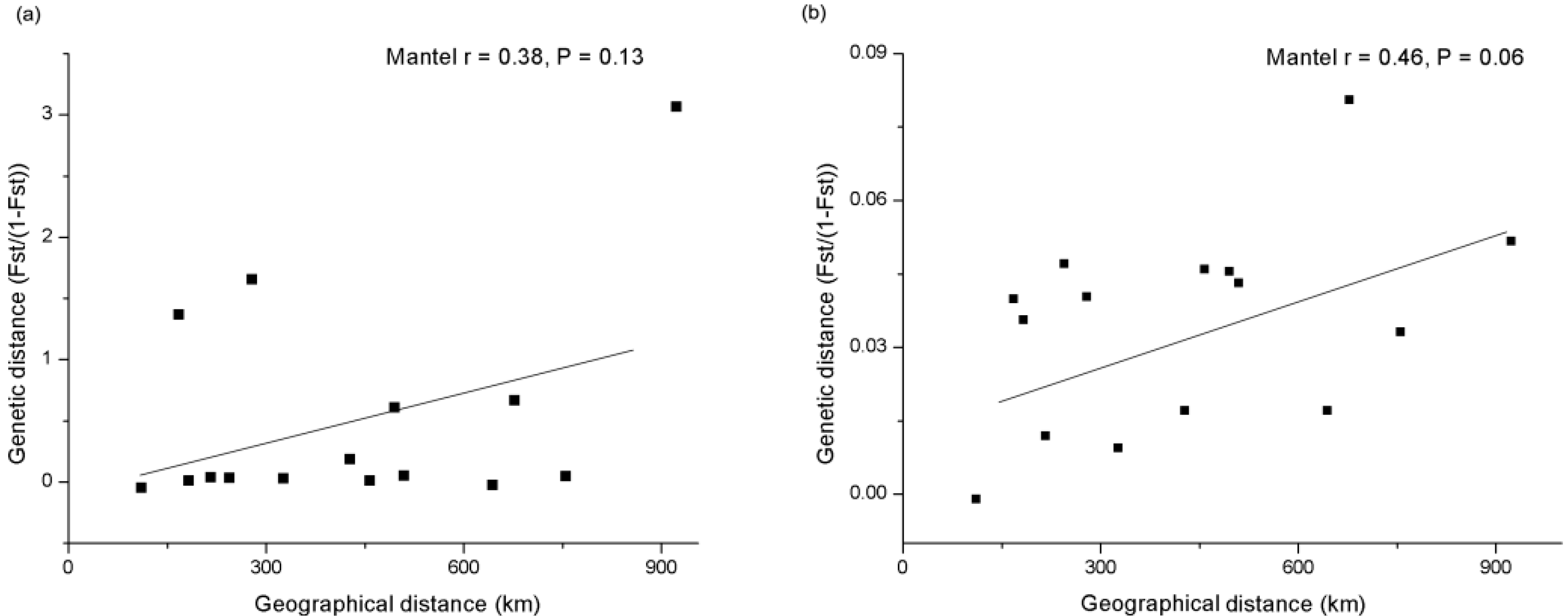
{kind=link}
{kind=link}
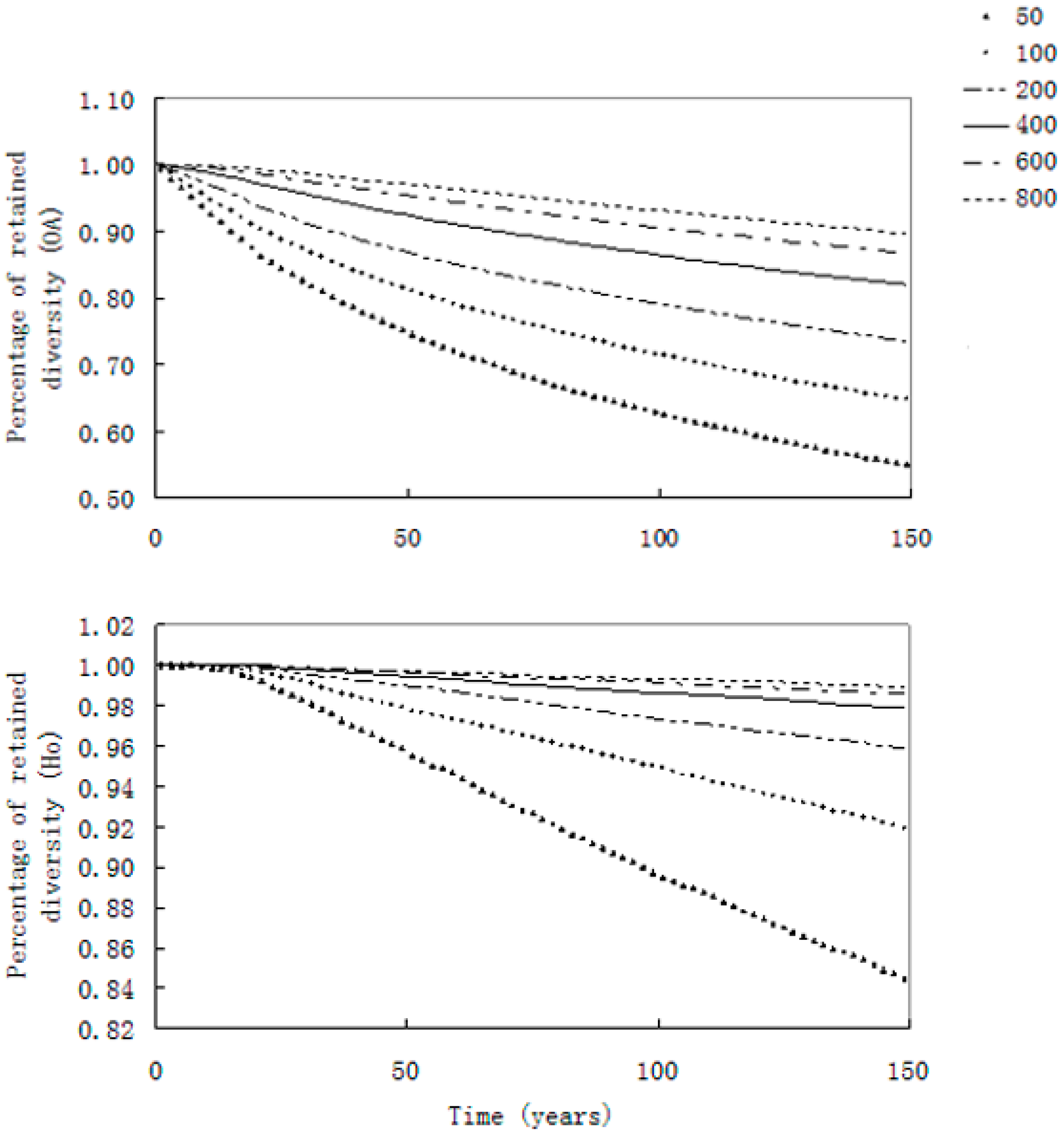{kind=link}
| Haplotype | Variable Site | Distribution of Haplotypes | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 1 | 2 | 3 | 3 | 3 | 5 | |||||||||||
| 3 | 0 | 8 | 1 | 0 | 0 | 6 | 0 | SH | TL | AQ | PY | EZHS | WH | HHPZ | DT | XCSS | Total | |
| 0 | 5 | 5 | 8 | 0 | 1 | 7 | 8 | |||||||||||
| NAACR-Hap1 | C | C | C | A | A | T | T | G | 0 | 3 | 0 | 30 | 4 | 0 | 3 | 3 | 12 | 55 |
| NAACR-Hap2 | T | . | . | . | . | . | . | . | 2 | 11 | 2 | 56 | 7 | 2 | 9 | 9 | 0 | 98 |
| NAACR-Hap3 | T | . | . | . | . | . | . | - | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| NAACR-Hap4 | . | A | . | G | . | . | . | . | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| NAACR-Hap5 | . | . | . | . | . | C | . | . | 0 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 0 | 5 |
| NAACR-Hap6 | T | . | T | . | . | . | . | . | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| NAACR-Hap7 | T | . | . | . | G | . | . | . | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| NAACR-Hap8 | T | . | . | . | . | . | C | . | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 5 |
| Locality | Microsatellite DNA | Mitochondrial DNA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| n | N | AR | Ho | He | Fis | n | h | π | |
| Total | 186 | 8.18 | 5.104 | 0.663 | 0.665 | −0.006 NS | 168 | 0.55 ± 0.03 | 0.0011 ± 0.0009 |
| TL | 17 | 5.36 | 4.617 | 0.699 | 0.642 | −0.019 NS | 16 | 0.51 ± 0.13 | 0.0009 ± 0.00076 |
| PY | 91 | 6.82 | 4.561 | 0.626 | 0.644 | 0.029 NS | 91 | 0.52 ± 0.04 | 0.0009 ± 0.00048 |
| EZHS | 15 | 5.27 | 5.230 | 0.649 | 0.631 | −0.054 NS | 13 | 0.65 ± 0.11 | 0.0016 ± 0.00129 |
| HHPZ | 14 | 5.45 | 5.271 | 0.663 | 0.702 | −0.061 NS | 14 | 0.56 ± 0.13 | 0.0013 ± 0.00079 |
| DT | 20 | 6.09 | 5.274 | 0.710 | 0.702 | −0.066 NS | 14 | 0.56 ± 0.13 | 0.0013 ± 0.00079 |
| XCSS | 16 | 5.27 | 4.582 | 0.696 | 0.649 | −0.083 NS | 12 | 0 | 0 |

2.2. Genetic Differentiation of the Yangtze Finless Porpoise
| TL | PY | EZHS | HHPZ | DT | XCSS | |
|---|---|---|---|---|---|---|
| TL | 0.032 | 0.027 | 0.032 | 0.032 | 0.669 *** | |
| PY | 0.038 *** | −0.004 | 0.001 | 0.001 | 0.466 *** | |
| EZHS | 0.030 * | 0.031 *** | −0.030 | −0.030 | 0.412 *** | |
| HHPZ | 0.018 | 0.042 *** | −0.003 | −0.077 | 0.495 *** | |
| DT | 0.027 * | 0.042 *** | 0.004 | −0.005 | 0.495 *** | |
| XCSS | 0.046 *** | 0.058 *** | 0.032 * | 0.031 * | 0.025 ** |


2.3. Simulations of the Evolution of Genetic Diversity in the Yangtze Finless Porpoise

3. Discussion
4. Experimental Section
4.1. Sample Collection and DNA Extraction
4.2. MtDNA Sequencing and Microsatellite Genotyping
4.3. Genetic Diversity Analysis
4.4. Genetic Differentiation and Population Structure Analysis
4.5. Simulations on the Evolution of Genetic Diversity
5. Conclusions and Conservation Implications
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Committee on Taxonomy. List of Marine Mammal Species and Subspecies. Society for Marine Mammalogy. Available online: http://www.marinemammalscience.org (accessed on 10 May 2010).
- Gao, A.; Zhou, K.Y. Geographical variation of external measurements and three subspecies of Neophocaena phocaenoides in Chinese waters. Acta Theriol. Sin. 1995, 15, 81–92. [Google Scholar]
- Wang, D. Population status, threats and conservation of the Yangtze finless porpoise. Chin. Sci. Bull. 2009, 54, 3473–3484. [Google Scholar]
- Wang, D.; Zhang, X.; Liu, R. Conservation status and its future of baiji and Yangtze finless porpoise in China. In Ecology and Environmental Protection of Large Irrigation Projects in Yangtze River in 21st Century; Hua, Z.L., Fu, B., Yang, Y., Eds.; Environmental Science Press: Beijing, China, 1998; pp. 218–226. [Google Scholar]
- Zhang, X.; Liu, R.; Zhao, Q.; Zhang, G.; Wei, Z.; Wang, X.; Yang, J. The population of finless porpoise in the middle and lower reaches of Yangtze River. Acta Theriol. Sin. 1993, 13, 260–270. [Google Scholar]
- Zhao, X.; Barlow, J.; Taylor, B.L.; Pitman, R.L.; Wang, K.; Wei, Z.; Stewart, B.S.; Turvey, S.T.; Akamatsu, T.; Reeves, R.R.; et al. Abundance and conservation status of the Yangtze finless porpoise in the Yangtze River, China. Biol. Conserv. 2008, 141, 3006–3018. [Google Scholar]
- Mei, Z.; Zhang, X.; Huang, S.L.; Zhao, X.; Hao, Y.; Zhang, L.; Qian, Z.; Zheng, J.; Wang, K.; Wang, D. The Yangtze finless porpoise: On an accelerating path to extinction? Biol. Conserv. 2014, 172, 117–123. [Google Scholar]
- Wang, D.; Turvey, S.T.; Zhao, X.; Mei, Z. Neophocaena asiaeorientalis ssp. asiaeorientalis. IUCN Red List of Threatened Species Version 2013.1. Available online: http://www.iucnredlist.org/ (accessed on 2 September 2013).
- Dong, S.Y. Studies on Distribution and Movement Pattern of Yangtze Finless Porpoise in Hukou Area by Acoustic Data Loggers. Master Dissertation, University of Chinese Academy of Sciences, Beijing, China, 2009. [Google Scholar]
- Zhang, X.Q. Population Ecology of Yangtze Finless Porpoise in Dongting Lake and the Adjacent Waters. Ph.D. Dissertation, University of Chinese Academy of Sciences, Beijing, China, 2011. [Google Scholar]
- Wei, Z.; Wang, D.; Zhang, X.; Zhao, Q.; Wang, K.; Kuang, X.A. Population size, behavior, movement pattern and protection of Yangtze finless porpoise at Balijiang section of the Yangtze River. Resour. Environ. Yangtze Basin 2002, 11, 427–432. [Google Scholar]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Kraaijeveld-Smit, F.J.L.; Beebee, T.J.C.; Griffiths, R.A.; Moore, R.D.; Schley, L. Low gene flow but high genetic diversity in the threatened Mallorcan midwife toad Alytes muletensis. Mol. Ecol. 2005, 14, 3307–3315. [Google Scholar]
- Lande, R.; Barrowclough, G. Effective population size, genetic variation, and their use in population management. In Viable Populations for Conservation; Soulé, M.E., Ed.; Cambridge University Press: New York, NY, USA, 1987; pp. 87–123. [Google Scholar]
- Simberloff, D. The contribution of population and community biology to conservation science. Annu. Rev. Ecol. Syst. 1988, 19, 473–511. [Google Scholar]
- Zheng, J.S.; Xia, J.H.; He, S.P.; Wang, D. Population genetic structure of the Yangtze finless porpoise (Neophocaena phocaenoides asiaeorientalis): Implications for management and conservation. Biochem. Genet. 2005, 43, 307–320. [Google Scholar]
- Yang, G.; Guo, L.; Bruford, M.W.; Wei, F.; Zhou, K. Mitochondrial phylogeography and population history of finless porpoises in Sino-Japanese waters. Biol. J. Linn. Soc. 2008, 95, 193–204. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A siulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar]
- Li, X.; Liu, Y.; Tzika, A.C.; Zhu, Q.; van Doninck, K.; Milinkovitch, M.C. Analysis of global and local population stratification of finless porpoises Neophocaena phocaenoides in Chinese waters. Mar. Biol. 2011, 158, 1791–1804. [Google Scholar]
- Rosel, P.E.; France, S.C.; Wang, J.Y.; Kocher, T.D. Genetic structure of harbour porpoise Phocoena phocoena populations in the northwest Atlantic based on mitochondrial and nuclear markers. Mol. Ecol. 1999, 8, S41–S54. [Google Scholar]
- Luca, M.; Andrew, W.; Emer, R.; Patricia, R.; Andrew, R.; Jamie, C.; Tom, C. Population structure of short-beaked common dolphins (Delphinus delphis) in the North Atlantic Ocean as revealed by mitochondrial and nuclear genetic markers. Mar. Biol. 2009, 156, 821–834. [Google Scholar]
- Hayano, A.; Amano, M.; Miyazaki, N. Phylogeography and population structure of the Dall’s porpoise, Phocoenoides dalli, in Japanese waters revealed by mitochondrial DNA. Genes Genet. Syst. 2003, 78, 81–91. [Google Scholar]
- Chen, L.; Bruford, M.W.; Xu, S.; Zhou, K.; Yang, G. Microsatellite variation and significant population genetic structure of endangered finless porpoises (Neophocaena phocaenoides) in Chinese coastal waters and the Yangtze River. Mar. Biol. 2010, 157, 1453–1462. [Google Scholar]
- Sellas, A.B.; Wells, R.S.; Rosel, P.E. Mitochondrial and nuclear DNA analyses reveal fine scale geographic structure in bottlenose dolphins (Tursiops truncatus) in the Gulf of Mexico. Conserv. Genet. 2005, 6, 715–728. [Google Scholar]
- Escorza-Trevino, S.; Archer, F.I.; Rosales, M.; Lang, A.; Dizon, A.E. Genetic differentiation and intraspecific structure of eastern tropical Pacific spotted dolphins, Stenella attenuata, revealed by DNA analyses. Conserv. Genet. 2005, 6, 587–600. [Google Scholar]
- Wang, D.; The Key Laboratory of Aquatic Biodiversity and Conservation of Chinese Academy of Sciences, Institute of Hydrobiology of Chinese Academy of Sciences, Wuhan 430072, China. unpublished work. 2014.
- Whitlock, M.C.; McCauley, D.E. Indirect measures of gene flow and migration: FST≠ 1/(4Nm+1). Heredity 1999, 82, 117–125. [Google Scholar]
- Mei, Z.; Huang, S.L.; Hao, Y.; Turvey, S.T.; Gong, W.; Wang, D. Accelerating population decline of Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis). Biol. Conserv. 2012, 153, 192–200. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Zheng, J.S.; Liao, X.L.; Tong, J.G.; Du, H.J.; Milinkovitch, M.C.; Wang, D. Development and characterization of polymorphic microsatellite loci in the endangered Yangtze finless porpoise (Neophocaena phocaenoides asiaeorientalis). Conserv. Genet. 2008, 9, 1007–1009. [Google Scholar]
- Zhou, Z.; Zheng, J.S.; Chen, M.M.; Zhao, Q.Z.; Wang, D. Genetic evaluation and development prognosis on ex situ conserved Yangtze finless porpoise living in Tian-E-Zhou National Natural Reserve. Acta Theriol. Sin. 2012, 36, 403–411. [Google Scholar]
- Chen, L.; Bruford, M.; Yang, G. Isolation and characterization of microsatellite loci in the finless porpoise (Neophocaena phocaenoides). Mol. Ecol. Notes 2007, 7, 1129–1131. [Google Scholar]
- Chen, L.; Yang, G. Development of tetranucleotide microsatellite loci for the finless porpoise (Neophocaena phocaenoides). Conserv. Genet. 2008, 9, 1033–1035. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.; Shipley, P. Micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar]
- Jeanmougin, F.; Thompson, J.D.; Gouy, M.; Higgins, D.G.; Gibson, T.J. Multiple sequence alignment with Clustal X. Trends Biochem. Sci. 1998, 23, 403–405. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar]
- Rousset, F. GENEPOP’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar]
- Goudet, J. FSTAT 2.9. 3.2, a Program to Estimate and Test Gene Diversities and Fixation Indices. Available online: http://www2.unil.ch/popgen/softwares/fstat.htm (accessed on 23 Febuary 2012).
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar]
- Ellegren, H. Microsatellite mutations in the germline: Implications for evolutionary inference. Trends Genet. 2000, 16, 551–558. [Google Scholar]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar]
- Luikart, G.; Cornuet, J.M. Empirical evaluation of a test for identifying recently bottlenecked populations from allele frequency data. Conserv. Biol. 1998, 12, 228–237. [Google Scholar]
- Wright, S. Evolution and the Genetics of Populations; University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Mantel, N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967, 27, 209–220. [Google Scholar]
- Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 1997, 145, 1219–1228. [Google Scholar]
- Franklin, I.R.; Frankham, R. How large must populations be to retain evolutionary potential? Anim. Conserv. 1998, 1, 69–73. [Google Scholar]
- Kuo, C.H.; Janzen, F.J. Bottlesim: A bottleneck simulation program for long-lived species with overlapping generations. Mol. Ecol. Notes 2003, 3, 669–673. [Google Scholar]
- Gao, A.; Zhou, K.Y. Growth and reproduction of three populations of finless porpoise, Neophocaena phocaenoides in Chinese waters. Aquat. Mamm. 1993, 19, 3–12. [Google Scholar]
- Hao, Y.; Wang, D.; Zhang, X. Review on breeding biology of Yangtze finless porpoise (Neophocaena phocaenoides asiaeorientalis). Acta Theriol. Sin. 2006, 26, 191–200. [Google Scholar]
- Frankel, O.H.; Soulé, M.E. Conservation and Evolution; Cambridge University Press: Cambridge, UK, 1981. [Google Scholar]
- Hedrick, P.W.; Kalinowski, S.T. Inbreeding depression in conservation biology. Annu. Rev. Ecol. Syst. 2000, 31, 139–162. [Google Scholar]
- Nieminen, M.; Singer, M.C.; Fortelius, W.; Schöps, K.; Hanski, I. Experimental confirmation that inbreeding depression increases extinction risk in butterfly populations. Am. Nat. 2001, 157, 237–244. [Google Scholar]
- Turvey, S.T.; Pitman, R.L.; Taylor, B.L.; Barlow, J.; Askamatsu, T.; Barrett, L.A.; Zhao, X.; Reeves, R.R.; Stewart, B.S.; Wang, K.; et al. First human-caused extinction of a cetacean species? Biol. Lett. 2007, 3, 527–540. [Google Scholar]
- Zhou, X.; Sun, F.; Xu, S.; Fan, G.; Zhu, K.; Liu, X.; Chen, Y.; Shi, C.; Yang, Y.; Huang, Z.; et al. Baiji genomes reveal low genetic variabiligy and new insights into secondary aquatic adaptations. Nat. Commun. 2013, 4, 1–6. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, M.; Zheng, J.; Wu, M.; Ruan, R.; Zhao, Q.; Wang, D. Genetic Diversity and Population Structure of the Critically Endangered Yangtze Finless Porpoise (Neophocaena asiaeorientalis asiaeorientalis) as Revealed by Mitochondrial and Microsatellite DNA. Int. J. Mol. Sci. 2014, 15, 11307-11323. https://doi.org/10.3390/ijms150711307
Chen M, Zheng J, Wu M, Ruan R, Zhao Q, Wang D. Genetic Diversity and Population Structure of the Critically Endangered Yangtze Finless Porpoise (Neophocaena asiaeorientalis asiaeorientalis) as Revealed by Mitochondrial and Microsatellite DNA. International Journal of Molecular Sciences. 2014; 15(7):11307-11323. https://doi.org/10.3390/ijms150711307
Chicago/Turabian StyleChen, Minmin, Jinsong Zheng, Min Wu, Rui Ruan, Qingzhong Zhao, and Ding Wang. 2014. "Genetic Diversity and Population Structure of the Critically Endangered Yangtze Finless Porpoise (Neophocaena asiaeorientalis asiaeorientalis) as Revealed by Mitochondrial and Microsatellite DNA" International Journal of Molecular Sciences 15, no. 7: 11307-11323. https://doi.org/10.3390/ijms150711307