2.1. Maternal and Fetal General Characteristics
There was no significant difference in body weight between the three groups before pregnancy. The mean body weight of the rats in AT1-AA group was slightly lower than that in control group one day before delivery, while there was no significant difference between AT1-AA + losartan group and control group (
Table 2). There was no significant difference in systolic blood pressure (SBP) between the three groups before pregnancy (
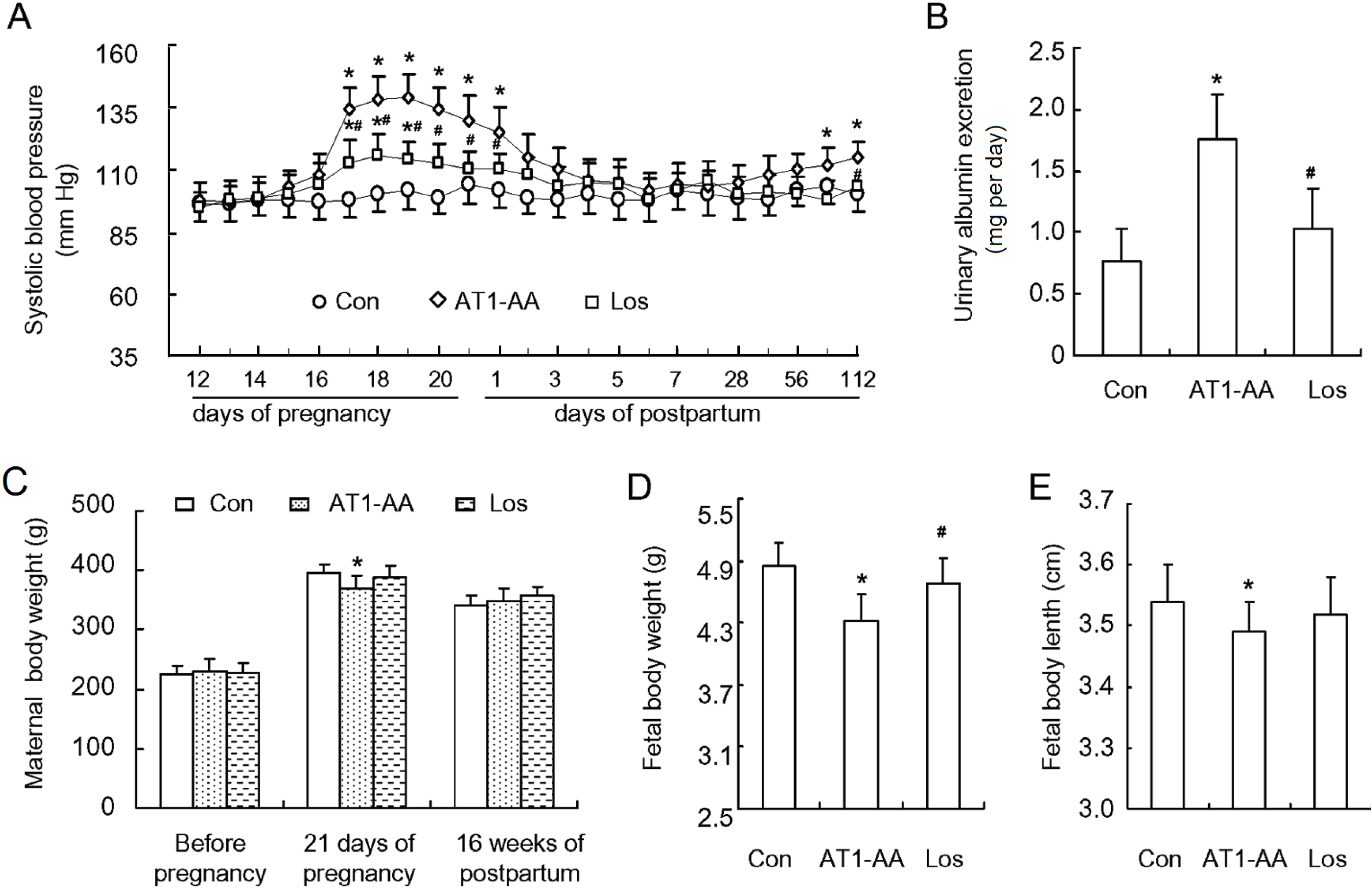
Figure 1A). SBP in AT1-AA group began rising at gestation day 16 (3 days after antibody injection), reached the peak (146 ± 12 mmHg) at gestation day 19, then declined gradually, and restored to normal at day 3 after delivery. However, SBP began rising gradually 12 weeks after delivery. Compared with control group, SBP in AT1-AA + losartan group was also elevated but the extent of SBP elevation was not so much as compared with AT1-AA group (
Figure 1A). In addition, SBP in AT1-AA + losartan group was not elevated after delivery. At the same time, the rats of AT1-AA group displayed a significantly raised urinary protein excretion (
Figure 1B). The gestational period of the three groups was between 21 and 22 days, showing no significant difference. Each rat bore 9–15 fetal rats, and there was no significant difference in the number of newborn rats between the three groups. However, the body weight and length of the newborn rats in AT1-AA group were significantly lower and shorter than those of control group. There was no significant difference in these parameters between AT1-AA + losartan and control groups (
Figure 1D,E). There was no significant difference in body weight between the three groups at 16 weeks postpartum.
Figure 1.
Affinity-purified AT1-AA (angiotensin II receptor type 1 autoantibody) induced an increase in SBP (systolic blood pressure) and proteinuria in pregnant rats. (A) SBP in pregnant rats after AT1-AA injection (1:640) at gestation day 13 in the presence or absence of losartan; (B) the ratio of urinary albumin/creatinine at gestation day 19; (C) maternal rat body weight (prior to, during and after pregnancy); (D) fetal rat body weight; and (E) fetal rat body length. Data represent the mean result from AT1-AA-injected rats, showing a significant increase in SBP and urinary protein. Con: control group; AT1-AA: AT1-AA group; Los: AT1-AA+losartan group. Data are expressed as means ± SD. (A–C): n = 6; (D,E): n = 32. * p < 0.05 vs. control group. # p < 0.05 vs. AT1-AA group.
Figure 1.
Affinity-purified AT1-AA (angiotensin II receptor type 1 autoantibody) induced an increase in SBP (systolic blood pressure) and proteinuria in pregnant rats. (A) SBP in pregnant rats after AT1-AA injection (1:640) at gestation day 13 in the presence or absence of losartan; (B) the ratio of urinary albumin/creatinine at gestation day 19; (C) maternal rat body weight (prior to, during and after pregnancy); (D) fetal rat body weight; and (E) fetal rat body length. Data represent the mean result from AT1-AA-injected rats, showing a significant increase in SBP and urinary protein. Con: control group; AT1-AA: AT1-AA group; Los: AT1-AA+losartan group. Data are expressed as means ± SD. (A–C): n = 6; (D,E): n = 32. * p < 0.05 vs. control group. # p < 0.05 vs. AT1-AA group.
2.2. LV (Left Ventricular) Function and Post-Ischemic Recovery
LV function and postischemic recovery using the Langendorff perfusion method. LV function was assessed in the isolated heart of the maternal rat exposed to either normal saline, AT1-AA, or AT1-AA + losartan at gestation day 13. The baseline left ventricular developed pressure (LVDP) and HR (heart rate) in AT1-AA group were slightly higher than those in control group, but there were no significant differences in left ventricular end-diastolic pressure (LVEDP), maximal rates of pressure rise or fall (dP/dt
max or dP/dt
min) and coronary flow at baseline levels. Meanwhile, the hemodynamic parameters of cardiac function of control group and AT1-AA + losartan group were not significantly different (
Table 1).
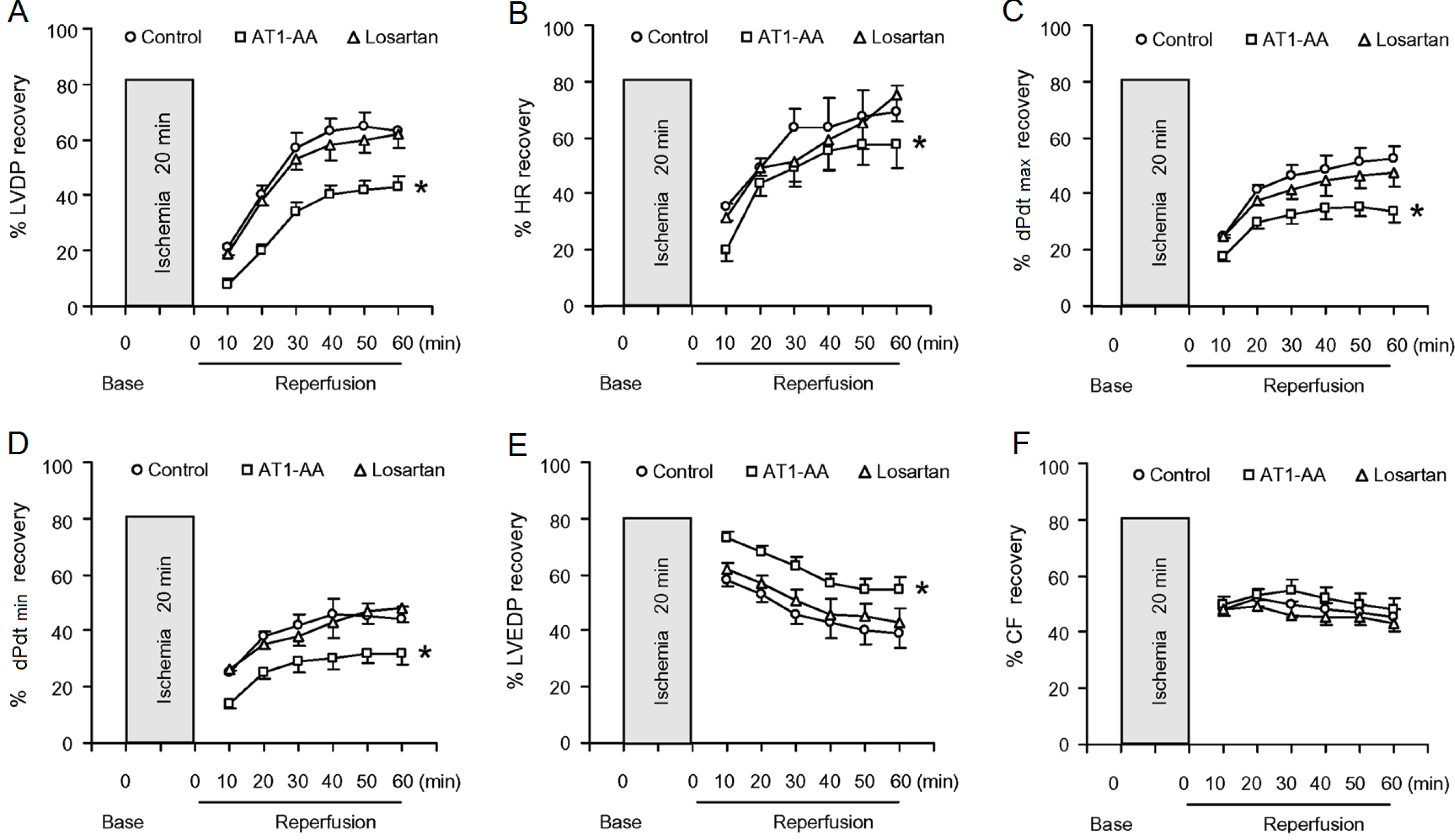
Figure 2 shows the effect of 20-min ischemia followed by 60-min reperfusion on LV function in the three groups. Cardiac contractility in rats gradually slowed down, or even completely stopped during 20-min ischemia. Although the reduced cardiac contractility gradually recovered following reperfusion, but had not complete recovery to the baseline value even 40 min after reperfusion. Compared with control group, there were significant decreases in post-ischemic recovery of the LVDP, and HR and +dp/dt
max in AT1-AA-treated group was delayed significantly, However, postischemic recovery of coronary flow was no significant difference between control group and AT1-AA-treated group. Ischemia reperfusion (IR) caused an injury to the heart and resulted in elevated LVEDP during reperfusion in isolated rat hearts. However, there was a significant increase in the IR-induced elevation of LVEDP in the AT1-AA-treated heart, as compared with the control heart (
Table 1). The functional parameters in AT1-AA + losartan group were significantly improved as compared with AT1-AA group.
Table 1.
Pre-ischemic LV (left ventricular) function parameters.
Table 1.
Pre-ischemic LV (left ventricular) function parameters.
| Parameter | Control | AT1-AA | AT1-AA + Losartan |
|---|
| LVDP (mmHg) | 116.6 ± 5.7 | 124.8 ± 6.2 | 118.2 ± 5.9 |
| HR (beats /min) | 309.2 ± 10.3 | 334.6 ± 13.2 * | 304.2 ± 12.3 # |
| LVEDP (mmHg) | 5.71 ± 0.83 | 5.81 ± 0.86 | 5.74 ± 0.72 |
| dp/dtmin (mmHg/s) | 2483 ± 106 | 2410 ± 156 | 2380 ± 146 |
| dp/dtmax (mmHg/s) | 3985 ± 519 | 4120 ± 634 | 3836 ± 533 |
| Coronary flow (mL/min) | 8.07 ± 0.68 | 7.83 ± 0.67 | 7.92 ± 0.61 |
Figure 2.
Effect of AT1-AA exposure on post-ischemic recovery of LV function (A–E) and coronary flow (F). Hearts obtained from the maternal rats exposed to either normal saline or AT1-AA (titer 1:640 0.1 mL/kg) at gestation days 13 and 14 were subjected to 20-min ischemia and 60-min reperfusion in the Langendorff preparation. LVDP: left ventricular developed pressure; HR: heart rate; dP/dtmax or dP/dtmin: the maximal rates of pressure rise or fall; LVEDP: left ventricular end diastolic pressure; CF: coronary flow; Con: control group; AT1-AA: AT1-AA group; Los: AT1-AA + losartan group. * p < 0.05 compared with control group for the entire curve. n = 6 per group.
Figure 2.
Effect of AT1-AA exposure on post-ischemic recovery of LV function (A–E) and coronary flow (F). Hearts obtained from the maternal rats exposed to either normal saline or AT1-AA (titer 1:640 0.1 mL/kg) at gestation days 13 and 14 were subjected to 20-min ischemia and 60-min reperfusion in the Langendorff preparation. LVDP: left ventricular developed pressure; HR: heart rate; dP/dtmax or dP/dtmin: the maximal rates of pressure rise or fall; LVEDP: left ventricular end diastolic pressure; CF: coronary flow; Con: control group; AT1-AA: AT1-AA group; Los: AT1-AA + losartan group. * p < 0.05 compared with control group for the entire curve. n = 6 per group.
2.3. Histopathological Change of the Myocardium
Although no significant difference in body weight was observed between the three groups at 16 weeks postpartum, the absolute and relative left ventricular weight (LVW) and LVW/BW (body weight) in AT1-AA group were significantly higher than those in control group (
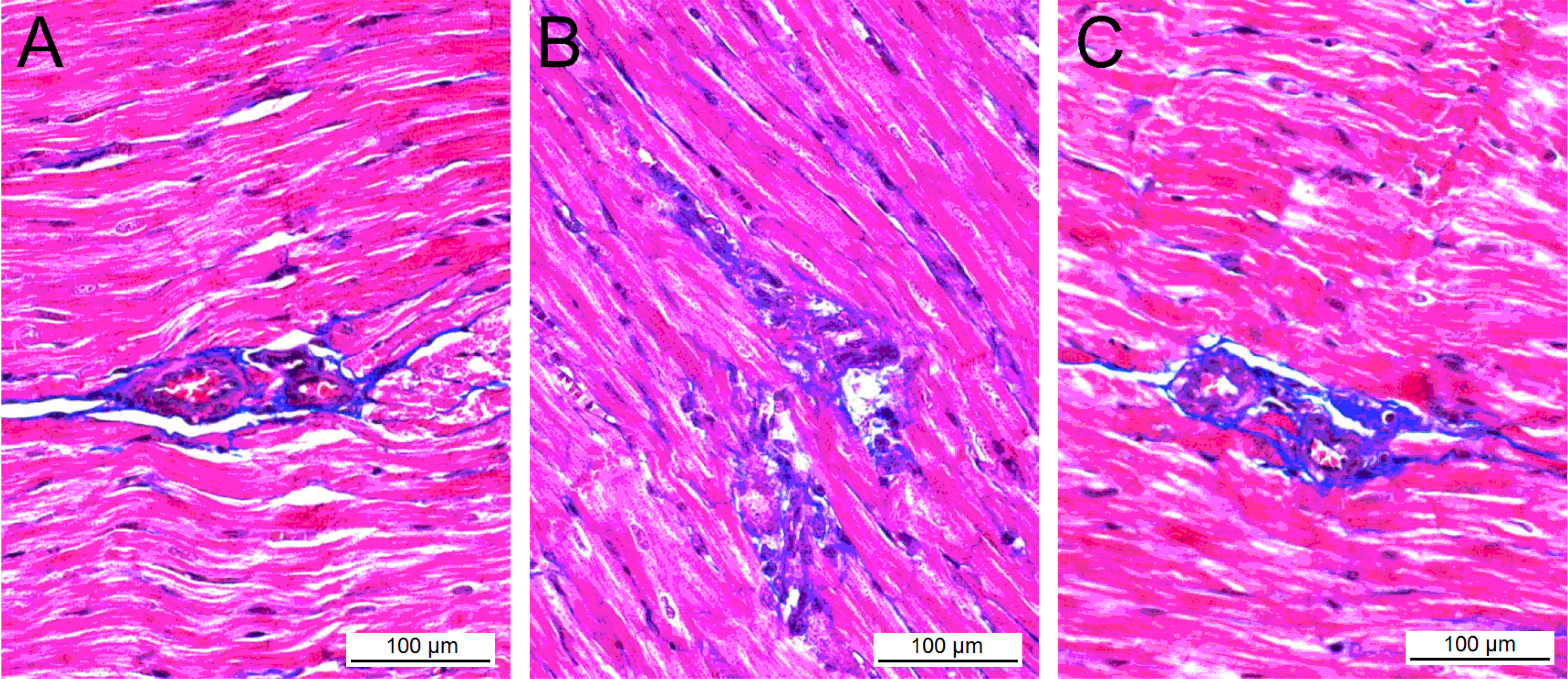
Table 2). The histological changes of the myocardial tissue were observed via light microscopy after Masson staining. The myocardial cells in control group were oval or short spindled, and arranged in neat dense rows with smaller intracellular spaces and tightly stained uniform nuclei with round and clear nucleoli, and no significantly abnormal change in the cellular structure was seen (
Figure 3A). The mean diameter of myocardial cells was 14.48 ± 2.79 μm, and the mean cross-sectional area (CSA) was 187.7 ± 32.9 μm
2. In AT1-AA group, the number of myocardial nuclei per microscopic field was significantly reduced as compared with control group (728.6 ± 69.2
vs. 962.7 ± 67.4,
p < 0.05), where cells were deranged and swollen, the nuclei were deeply stained, and cytolymph and necrosis were observed in the myocardial tissue (
Figure 3B).
Table 2.
Heart weight index and morphometric analysis of LV sarcomeres and mitochondria of rats. LVW: left ventricular weight; BW: body weight.
Table 2.
Heart weight index and morphometric analysis of LV sarcomeres and mitochondria of rats. LVW: left ventricular weight; BW: body weight.
| Parameter | Control | AT1-AA | AT1-AA + Losartan |
|---|
| BW (g) | 369 ± 11 | 358 ± 12 | 374 ± 14 |
| LVW (mg) | 689 ± 12 | 798 ± 14 * | 708 ± 15 # |
| LVW/BW (mg/g) | 1.87 ± 0.08 | 2.23±0.12 * | 1.94±0.09 # |
| Infarct size (%) | 17.6 ± 1.2 | 28.4 ± 1.8 * | 19.7 ± 1.6 # |
| Cardiomyocyte diameter (μm) | 14.48 ± 2.79 | 18.13 ± 4.27 | 15.13 ± 3.12 |
| Cross-sectional area (μm2) | 187.7 ± 32.9 | 287.6 ± 59.4 * | 201.2 ± 40.1 # |
| Myocardial nuclei number | 962.7 ± 67.4 | 728.6 ± 69.2 * | 947.6 ± 74.3 # |
Figure 3.
Masson’s trichrome stain of the heart in control group (A), AT1-AA group (B), and AT1-AA + losartan group (C). Where blue = fibrous collagen, and red = cardiomyocytes. (A) The comparative fibrosis of control rats (B) AT1-AA treated rats showing an increased amount of collagen deposition compared with control rats (p < 0.05); and (C) Less collagen accumulation in the heart of AT1-AA + losartan treated rats, compared with AT1-AA group (p < 0.05); representative histological sections.
Figure 3.
Masson’s trichrome stain of the heart in control group (A), AT1-AA group (B), and AT1-AA + losartan group (C). Where blue = fibrous collagen, and red = cardiomyocytes. (A) The comparative fibrosis of control rats (B) AT1-AA treated rats showing an increased amount of collagen deposition compared with control rats (p < 0.05); and (C) Less collagen accumulation in the heart of AT1-AA + losartan treated rats, compared with AT1-AA group (p < 0.05); representative histological sections.
The CSA of myocardial cells in AT1-AA group was 287.6 ± 59.4 μm
2, which were significantly higher than those of control group (
p < 0.01). No significant damage to the myocardial structure was observed in AT1-AA + losartan group, the mean diameter and CSA of myocardial cells compared with the control group did not differ significantly (
p > 0.05) (
Table 2).
2.4. Effects of AT1-AA (Angiotensin II Receptor Type 1 Autoantibody) on Myocardial Fibrosis
Myocardial fibrosis is related to excessive collagen synthesis, and hydroxyproline measurement can accurately reflect the amount of collagen in the tissue, by which we can indirectly understand the extent of myocardial fibrosis. The content of hydroxyproline in the myocardium of AT1-AA group was significantly higher than that in control group (
p < 0.01), and there was less collagen accumulation in AT1-AA + losartan group compared with AT1-AA group, but there is no statistically significant difference (
Table 3).
Table 3.
Effects of AT1-AA on interstitial collagen volume fraction and peri-vascular collagen area-to-luminal area ratio of the LV.
Table 3.
Effects of AT1-AA on interstitial collagen volume fraction and peri-vascular collagen area-to-luminal area ratio of the LV.
| Parameter | Control | AT1-AA | AT1-AA + Losartan |
|---|
| hydroxyproline content (mg/g) | 2.14 ± 0.51 | 2.98 ± 0.76 * | 2.47 ± 0.54 |
| collagen content (mg/g) | 15.82 ± 2.46 | 22.55 ± 4.23 * | 18.47 ± 2.59 # |
| collagen volume fraction (%) | 3.72 ± 0.48 | 14.22 ± 1.54 * | 5.79 ± 0.64 # |
| perivascular collagen area-to-luminal area ratio (%) | 5.76 ± 0.94 | 16.14 ± 2.12 * | 8.11 ± 1.13 # |
Cardiac fibrosis was observed in AT1-AA group, presenting as a diffuse, small, patchy and non-uniform pattern, where the collagen network structure in the interstitial and peri-vascular areas was destroyed and disorganized (
Figure 3B). When compared with control group, the collagen volume fraction and perivascular collagen area/luminal area in AT1-AA group were markedly increased (
p < 0.01 and
p < 0.05, respectively). However, the collagen volume fraction and perivascular collagen area/luminal area in AT1-AA + losartan group were significantly lower than those in AT1-AA (
p < 0.01 and
p < 0.05, respectively), suggesting that Iosartan improved the AT1-AA-induced myocardial fibrosis (
Figure 3C).
2.5. Ultrastructural Changes of the Myocardium
Microscopic observation on the LV ultrastructure in control group showed that myocardial fibers were arranged in neat rows, with bright bands, dark bands, Z lines and clear intercalated discs. The mean length of sarcomeres was 1.63 ± 0.07 μm. Mitochondria between the myocardial fibers were mostly oval in a beaded arrangement with clear lamellae (
Figure 4A,D). In AT1-AA group, myocardial cells were wrinkled, myofibers were ruptured, and sarcomeres were blurred, with dissolution of sarcomeric organization and disruption of the intercalated disc. The length of sarcomeres averaged about 1.85 ± 0.11 μm. The number of mitochondria was decreased, and mitochondria were densely arranged. The cytoplasm was concentrated with more vacuoles inside. The mitochondrial ridge and inner-membrane fusion disappeared partially or completely (
Figure 4B,E). In AT1-AA + losartan group, myocardial fibers were neatly arranged with clear intercalated discs and clear sarcomeres, the length of which averaged 1.68 ± 0.08 μm; the number of mitochondria gradually increased (
Figure 4C,F), (
Table 4).
Figure 4.
Transmission electron microscopy of myocardium after 20 min of ischemia followed by 60 min of reperfusion in control group (A,D), AT1-AA group (B,E), and AT1-AA + losartan group (C,F). (A,D) Normal nuclei and mitochondria in control group; (B,E) The mitochondria are swollen, partly disrupted, and contain flocculent densities; (C,F) Sarcomeres are moderately relaxed and mitochondria are swollen in AT1-AA + losartan group. Original magnification (A,B) 8400×; (C–F) 22,500×.
Figure 4.
Transmission electron microscopy of myocardium after 20 min of ischemia followed by 60 min of reperfusion in control group (A,D), AT1-AA group (B,E), and AT1-AA + losartan group (C,F). (A,D) Normal nuclei and mitochondria in control group; (B,E) The mitochondria are swollen, partly disrupted, and contain flocculent densities; (C,F) Sarcomeres are moderately relaxed and mitochondria are swollen in AT1-AA + losartan group. Original magnification (A,B) 8400×; (C–F) 22,500×.
Table 4.
Morphometric analysis of rat LV sarcomeres and mitochondria.
Table 4.
Morphometric analysis of rat LV sarcomeres and mitochondria.
| Parameter | Control | AT1-AA | AT1-AA + Losartan |
|---|
| Sarcomere width (μm) | 1.63 ± 0.07 | 1.85 ± 0.11 * | 1.68 ± 0.08 # |
| Sarcomere length (μm) | 0.47 ± 0.06 | 0.74 ± 0.08 * | 0.52 ± 0.06 # |
| Volume density (%) | 33.41 ± 3.62 | 25.22 ± 4.21 * | 38.75 ± 3.66 # |
| Numerical density (/μm3) | 0.89 ± 0.11 | 0.75 ± 0.16 | 0.92 ± 0.15 |
| Mean volume (μm3) | 0.67 ± 0.08 | 1.02 ± 0.08 * | 0.73 ± 0.08 # |
2.6. Discussion
The volume of blood increases by more than 30% during pregnancy because of the establishment of fetal placental circulation and change of the endocrine system. In addition, enlargement of the uterus in the late stage of pregnancy pushes the diaphragm upward, causing upward and left anterior displacement of the heart, resulting in mild twisting of the major cardiac vessels, which further increases the burden of the maternal heart. The cardiac structure and function of pregnant women would be seriously affected when complications of hypertension, PE and diabetes occur. PE (eclampsia) constitutes the greatest threat to the heart of pregnant women both in developing and developed countries, and is also generally recognized as the main cause of premature birth, fetal and maternal death [
1]. The study by Melchiorre
et al. [
15] study demonstrate that preterm PE is strongly associated with the persistence of LV dysfunction/hypertrophy at one year postpartum follow-up and the risk of development of essential hypertension within two years. As the pathogenesis of PE remains elusive, there is no effective means for the prevention and treatment of the condition at present.
Ample studies have demonstrated that ventricular remodeling is closely associated with the abnormal RAS. Angiotensin II is the main component of the RAS. It induces hypertrophy, apoptosis and fibrosis of myocardial cells and causes other pathological changes, resulting in ventricular remodeling. AT1R inhibitors can effectively prevent or even reverse hypertrophy and fibrosis of myocardial cells, thus improving the cardiac function [
16]. It is interesting to find that RAS components in the plasma of normal pregnant women are elevated significantly, while angiotensinogen, plasma renin activity and Ang II level in the plasma of preeclamptic pregnant women are significantly lower than those in normal pregnant women [
9]. AT1-AA is a newly discovered immunoglobulin G (IgG) in the plasma of preeclamptic pregnant women. It can act on the second extracellular loop of AT1R to produce an Ang II agonist-like effect and pathologic impairment. Siddiqui
et al. [
17] found that plasma AT1-AA levels were elevated markedly in more than 95% of preeclamptic patients. Wenzel
et al. [
18] reported that AT1-AA could increase the sensitivity of pregnant rats to Ang II Combination use of AT1-AA and Ang II induced hypertension, proteinuria, intrauterine growth retardation and arteriolosclerosis in the uteroplacental unit, but use of Ang II alone did not induce these symptoms. Ample studies have demonstrated that AT1-AA participates in the pathogenesis of preeclampsia by inducing the expression of multiple cytokines closely associated with the onset of the disease [
10,
19].
Our previous studies showed that AT1-AA could induce hypertrophy and apoptosis of isolated myocardial cells in lactating rats [
13], and antibodies produced by immunization of rats with the second extracellular loop of AT1R could induce apoptosis and hypertrophy of myocardial cells and remodeling of the myocardium, leading to hypertension [
13]. The result of the presents study showed that pregnant rats injected with AT1-AA at gestation day 13 developed hypertension, and at gestational day 18 developed proteinuria, and the body weight and length of the newborn rats were lower and shorter. These manifestations are very similar to the symptoms of human preeclampsia, indicating that AT1-AA could also induce the occurrence of preeclampsia. At 16 weeks postpartum, the LVW and LVWI in the rats of AT1-AA group increased significantly; the CSA of myocardial cells was increased, and there were large amounts of collagen fiber deposition around the myocardial interstitial and vessels, indicating that the abnormally expressed AT1-AA in the plasma of preeclamptic women could lead to cardiac remodeling. This damage to myocardial cells was at the expense of increasing the size of myocardial cells, thus decreasing the functional reserve of the heart, and therefore the cardiac function can only meet the requirements of human resting levels. Under the condition of myocardial ischemia, the cardiac pumping function becomes insufficient. In addition, re-arrangement of myocardial cells is likely to trigger arrhythmia. As ventricular remodeling is a progressive process, LV dilatation, dysfunction and heart failure are prone to occur without effective prevention and treatment [
20]. Studies [
21] have shown that a hypertrophic heart has poor tolerance to ischemia, and is slow or unable to restore its function completely. It was observed in our study that the rat LVW, LVWI, CSA, collagen concentration and collagen area around blood vessels were all markedly decreased in AT1-AA + losartan group, and these changes were significantly different from those in rats treated with AT1-AA group (
p < 0.05 or
p < 0.01), indicating that losartan could delay or block the occurrence of AT1-AA induced myocardial hypertrophy and elevation of the collagen content in myocardial interstitial cells, thus attenuating or improving ventricular remodeling. Therefore, we postulate that AT1-AA existing in the plasma of pregnant women could induce cellular hyperplasia, which may be an important mechanism underlying the increased susceptibility of the postpartum heart to IRI.
Histological study of the rat heart using light microscopy showed spotty and patchy necroses in myocardial cells in the rats of AT1-AA group. These necrotic lesions were mainly located under the endocardium, and partly across the full length of the ventricular muscular wall. They had been replaced by the proliferative fibrous tissue. Transmission electron microscopic examination of the myocardial ultrastructure showed that the myocardial cells had undergone degenerative, necrotic and hyperplasia-like changes with edematous interstitial and large amounts of collagen; part of the myofilaments were ruptured, and mitochondria were swollen without observing crests and with vacuolar degeneration observed. After the use of losartan, the ultrastructure of the myocardial cells was improved significantly as compared with that of the AT1-AA group. The structure of myocardial cells is the footstone of cardiac function. Damage to myocardial cells will decrease the cardiac pumping function, resulting in various cardiac diseases. Our previous study demonstrated that AT1-AA could enter the fetal rat via the placenta and induce the generation of large amounts of TNF-α, thus promoting apoptosis of myocardial cells of the fetal rat and thus increasing the susceptibility of the heart of these offspring to IRI 10 weeks postpartum. Xia
et al. [
10,
11] found that the pathogenesis of AT1-AA-induced preeclamsia was closed associated with TNF-α. They found that the TNF-α concentration was increased markedly in the plasma of AT1-AA treated animals, and the use of TNF-α inhibitors could effectively attenuate AT1-AA-induced hypertension, preeclampsia and placental apoptosis. The result of the present study showed that SBP and HR were increased in AT1-AA group 16 weeks postpartum. The
in vitro cardiac perfusion experiment showed that the LV function restored more slowly after myocardial ischemia, and the myocardial infarct area was also increased significantly, which may be due to the AT1-AA-induced generation of TNF-α, causing apoptosis of myocardial cells.
The NLRP3 (nucleotide-binding domain leucine-rich repeat containing family, pyrin domain containing 3) inflammasome complex, assembled in response to microbial components or endogenous danger signals, triggers caspase-1-mediated maturation and secretion of IL-1β and IL-18 processing, a key step in the innate immune response [
22]. Several studies [
23,
24] have shown that cardiac myocytes and cardiac fibroblasts express NLRP3, which is involved in the pathogenesis of myocardial ischemia-reperfusion injury, while the NLRP3 inflammasome is up-regulated in myocardial fibroblasts after myocardial infarction, and may be a significant contributor to infarct size development during IR. NLRP3 agonists that have been tested trigger the production of reactive oxygen species (ROS) [
25]. ROS production results in NLRP3 inflammasome activation through release of the ROS-sensitive NLRP3 ligand thioredoxin-interacting protein from its inhibitor thioredoxin [
26]. Parrish
et al. [
27] suggest that AT1-AA through activation of NADPH oxidase could contribute to ROS production and inflammatory responses in PE. Therefore, our future plan is to further study the role and participation of NLRP3 inflammasome in AT1-AA-induced increases in cardiac susceptibility to IRI.



{kind=link}
{kind=link}
{kind=link}
{kind=link}


