Overcoming Hypoxic-Resistance of Tumor Cells to TRAIL-Induced Apoptosis through Melatonin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
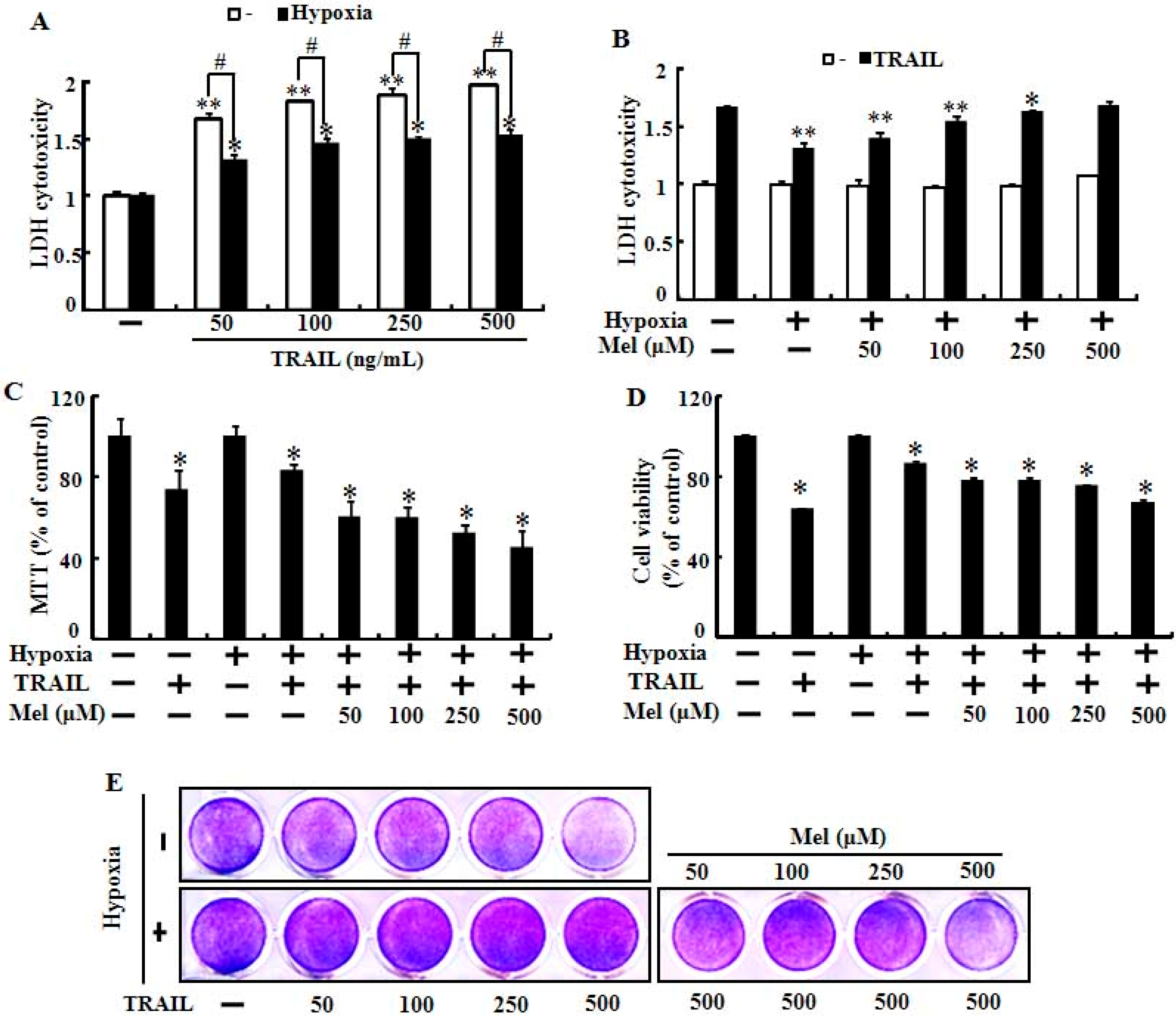
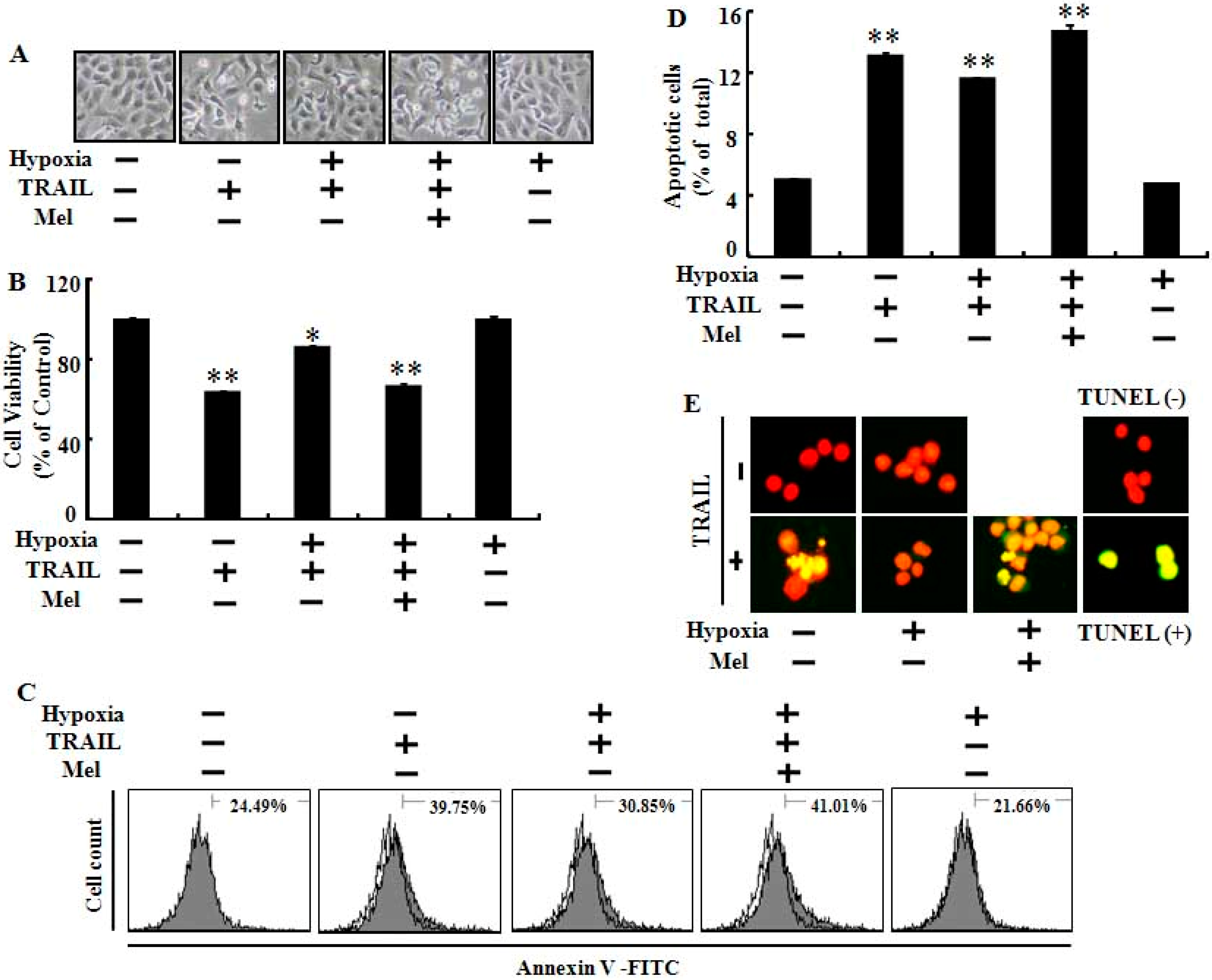
2.1. Melatonin Enhanced TRAIL (Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand)-Induced Human Lung Cancer Cell Death under Hypoxic Conditions
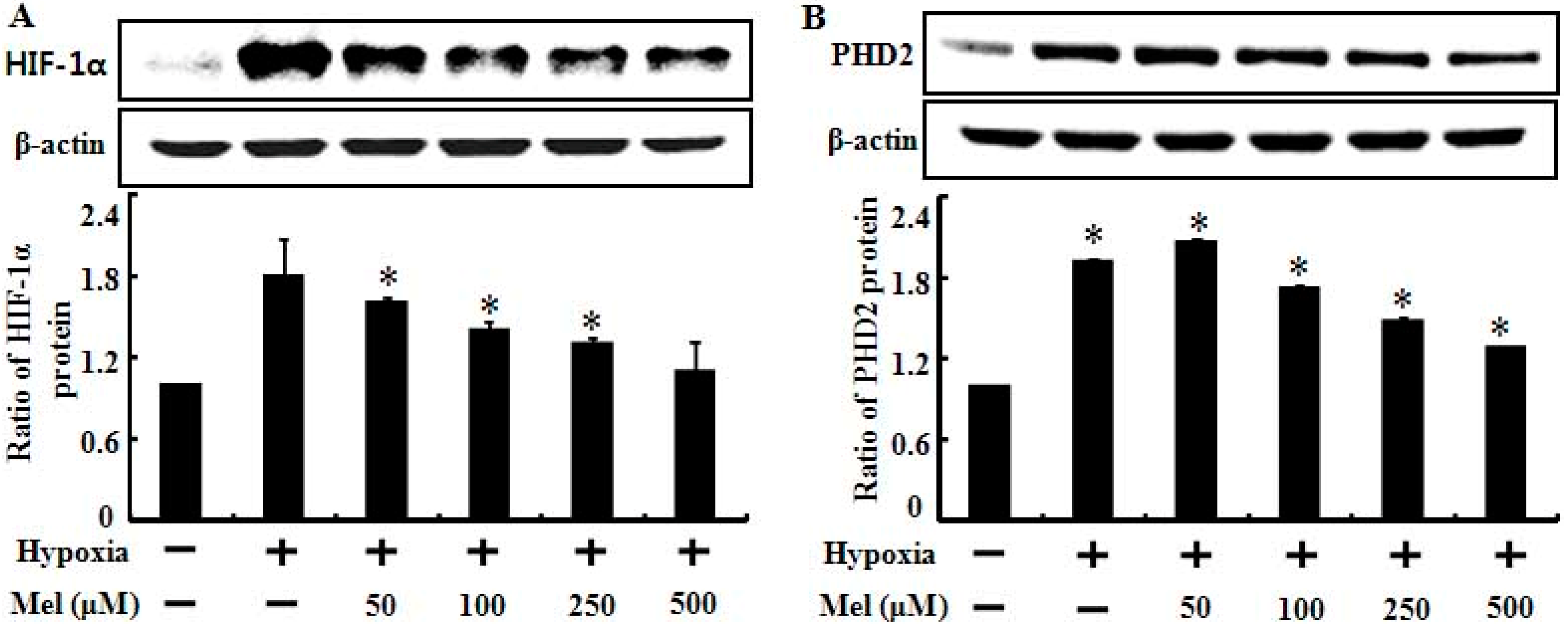
2.2. Melatonin Down-Regulates Hypoxia-Related Protein Expression



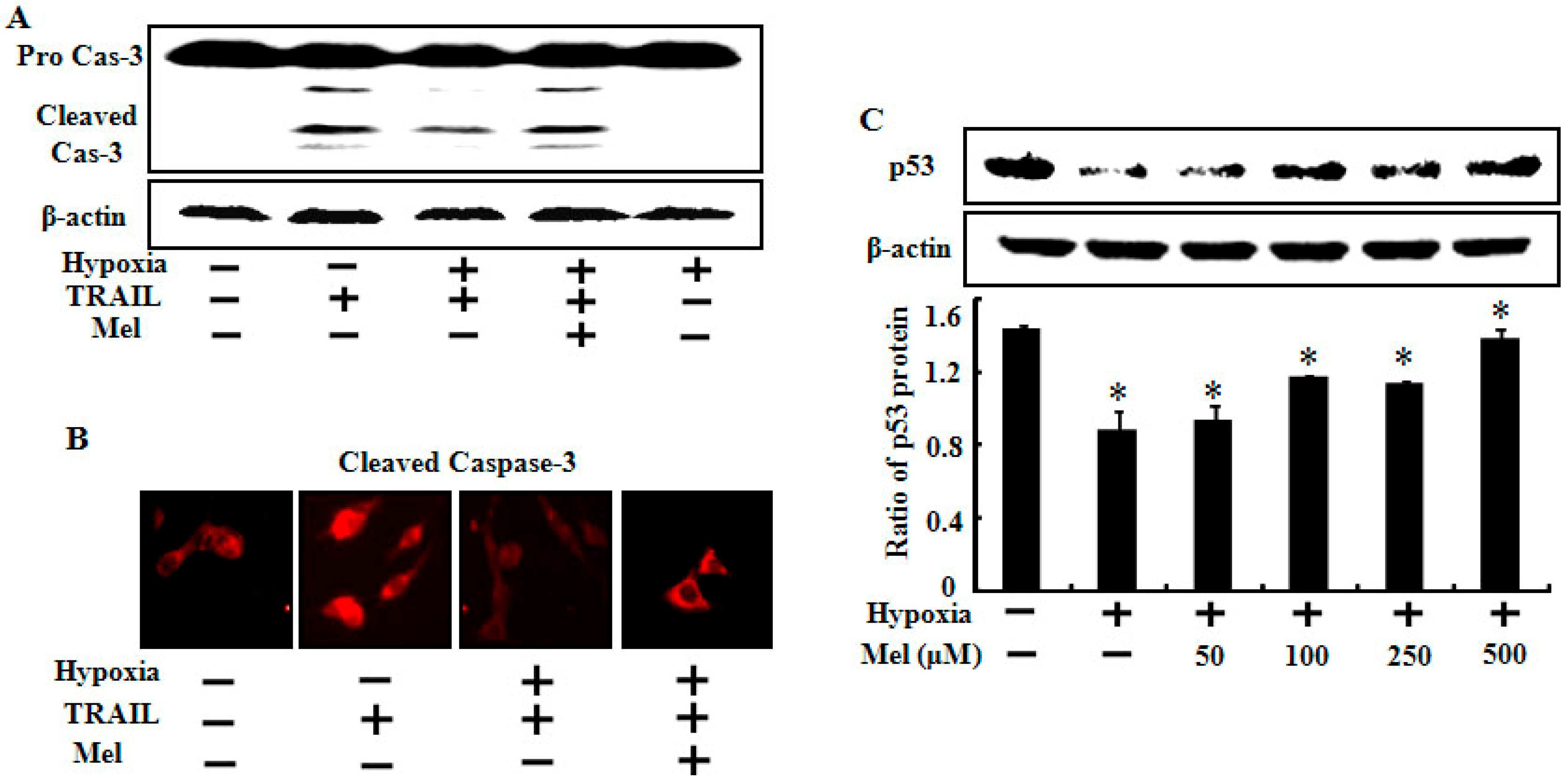
2.3. Melatonin Up-Regulates Pro-Apoptotic Signals Mediated by Hypoxia

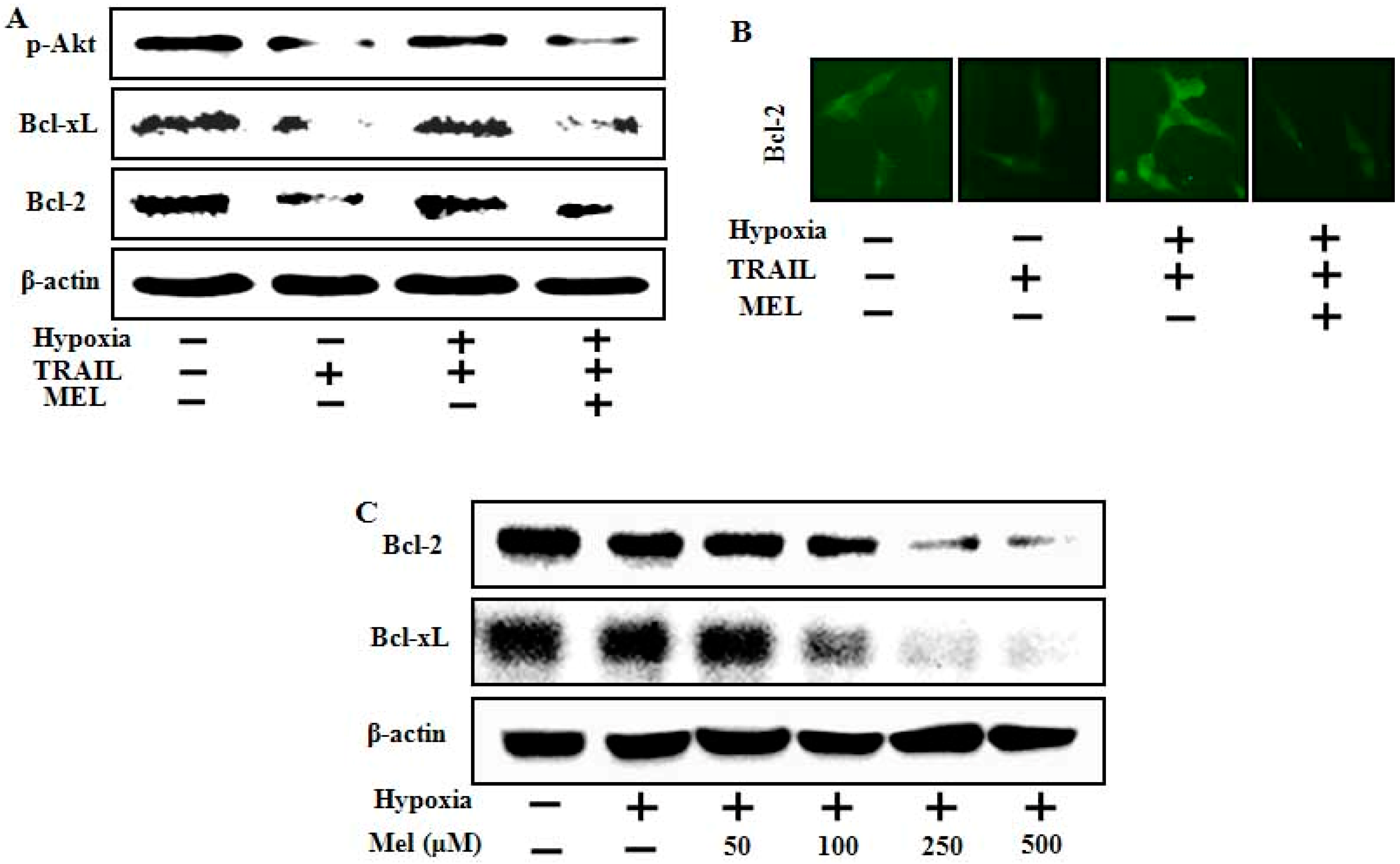
2.4. Melatonin Down-Regulates Anti-Apoptotic Signals Mediated by Hypoxia

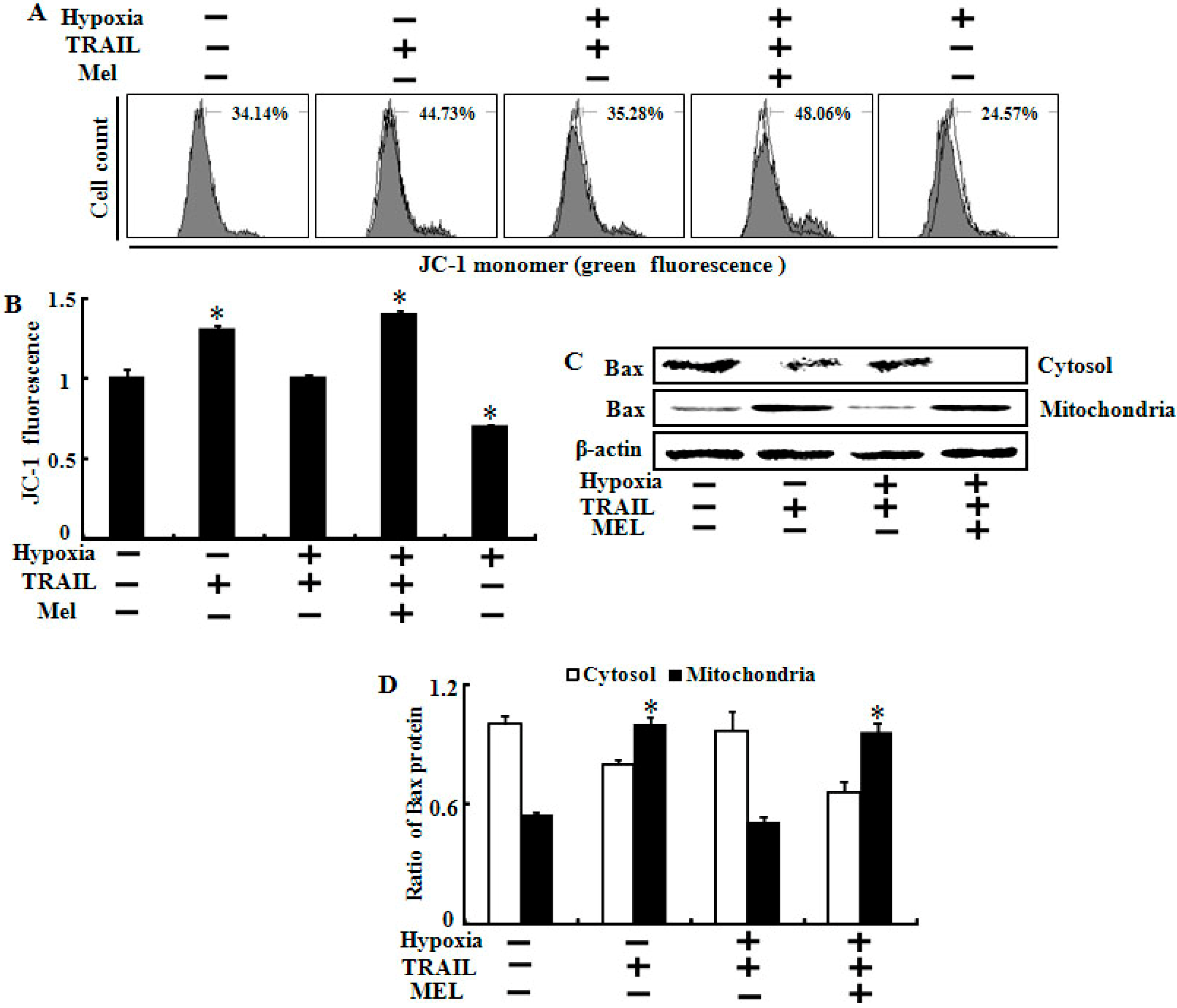
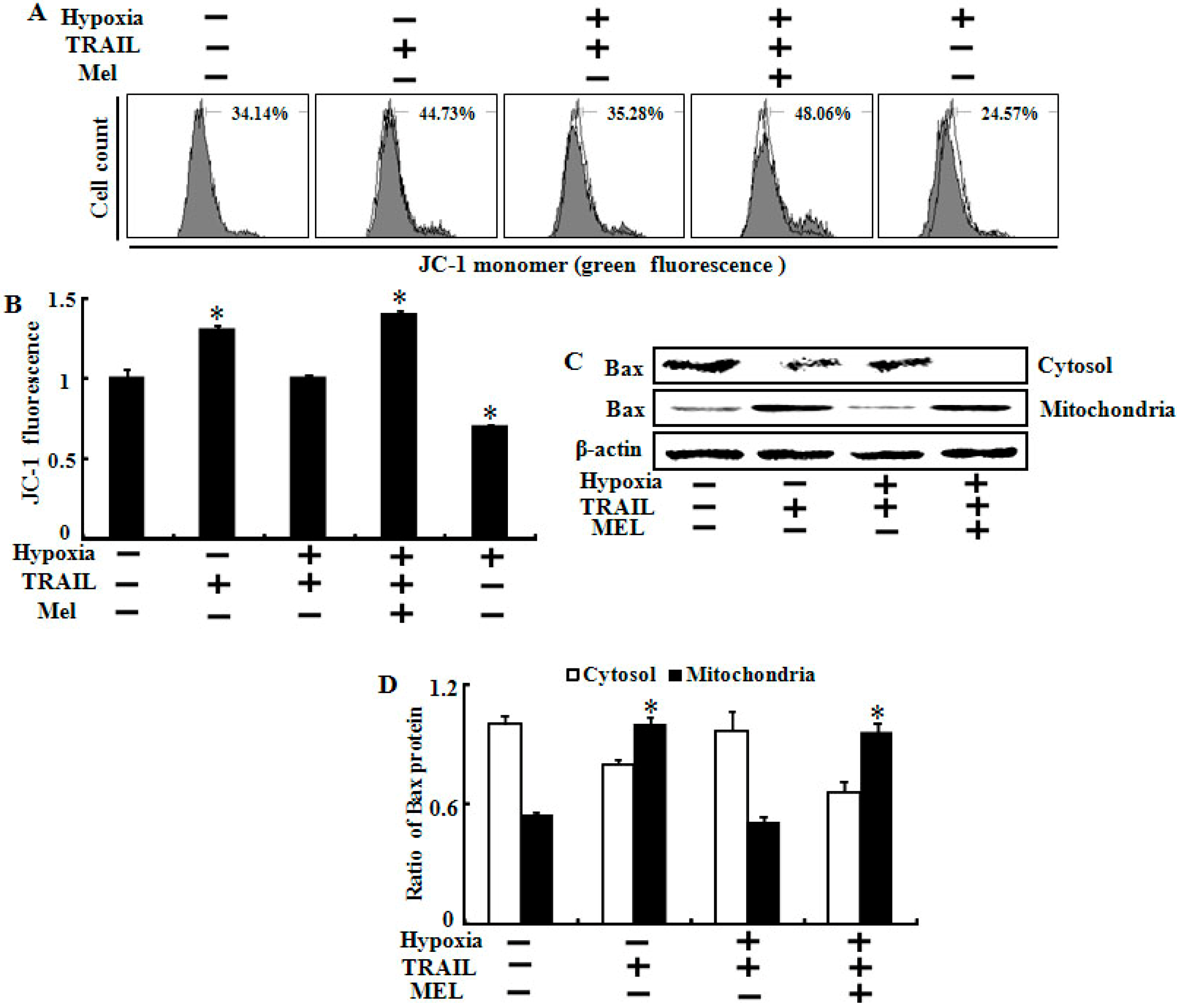
2.5. Melatonin Inhibited Hypoxia-Mediated Regulation of Mitochondrial Dysfunction and Bax Translocation
2.6. Discussion
3. Experimental Section
3.1. Cell Culture and Reagents
3.2. Hypoxic Conditions
3.3. Lactate Dehydrogenase (LDH) Assay
3.4. Crystal Violet Assay
3.5. MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Assay
3.6. Annexin V Assay
3.7. Terminal Uridine Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
3.8. Western Blot Analyses
3.9. Cellular Fractionation
3.10. Immunofluorescent Staining
3.11. Mitochondrial Transmembrane Potential (MTP) Assay
3.12. Statistical Evaluation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hardeland, R.; Tan, D.X.; Reiter, R.J. Kynuramines, metabolites of melatonin and other indoles: The resurrection of an almost forgotten class of biogenic amines. J. Pineal Res. 2009, 47, 109–126. [Google Scholar] [CrossRef]
- Simko, F.; Pechanova, O. Potential roles of melatonin and chronotherapy among the new trends in hypertension treatment. J. Pineal Res. 2009, 47, 127–133. [Google Scholar]
- Veneroso, C.; Tunon, M.J.; Gonzalez-Gallego, J.; Collado, P.S. Melatonin reduces cardiac inflammatory injury induced by acute exercise. J. Pineal Res. 2009, 47, 184–191. [Google Scholar] [CrossRef]
- Nosjean, O.; Nicolas, J.P.; Klupsch, F.; Delagrange, P.; Canet, E.; Boutin, J.A. Comparative pharmacological studies of melatonin receptors: Mt1, mt2 and mt3/qr2. Tissue distribution of mt3/qr2. Biochem. Pharmacol. 2001, 61, 1369–1379. [Google Scholar] [CrossRef]
- Reiter, R.J.; Paredes, S.D.; Manchester, L.C.; Tan, D.X. Reducing oxidative/nitrosative stress: A newly-discovered genre for melatonin. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 175–200. [Google Scholar] [CrossRef]
- Peyrot, F.; Ducrocq, C. Potential role of tryptophan derivatives in stress responses characterized by the generation of reactive oxygen and nitrogen species. J. Pineal Res. 2008, 45, 235–246. [Google Scholar] [CrossRef]
- Gonzalez, A.; del Castillo-Vaquero, A.; Miro-Moran, A.; Tapia, J.A.; Salido, G.M. Melatonin reduces pancreatic tumor cell viability by altering mitochondrial physiology. J. Pineal Res. 2011, 50, 250–260. [Google Scholar] [CrossRef]
- Buscemi, N.; Vandermeer, B.; Hooton, N.; Pandya, R.; Tjosvold, L.; Hartling, L.; Vohra, S.; Klassen, T.P.; Baker, G. Efficacy and safety of exogenous melatonin for secondary sleep disorders and sleep disorders accompanying sleep restriction: Meta-analysis. BMJ 2006, 332, 385–393. [Google Scholar] [CrossRef]
- Comperatore, C.A.; Krueger, G.P. Circadian rhythm desynchronosis, jet lag, shift lag, and coping strategies. Occup. Med. 1990, 5, 323–341. [Google Scholar]
- Cardinali, D.P.; Srinivasan, V.; Brzezinski, A.; Brown, G.M. Melatonin and its analogs in insomnia and depression. J. Pineal Res. 2012, 52, 365–375. [Google Scholar] [CrossRef]
- Vijayalaxmi; Thomas, C.R., Jr.; Reiter, R.J.; Herman, T.S. Melatonin: From basic research to cancer treatment clinics. J. Clin. Oncol. 2002, 20, 2575–2601. [Google Scholar] [CrossRef]
- Park, S.Y.; Jang, W.J.; Yi, E.Y.; Jang, J.Y.; Jung, Y.; Jeong, J.W.; Kim, Y.J. Melatonin suppresses tumor angiogenesis by inhibiting HIF-1α stabilization under hypoxia. J. Pineal Res. 2010, 48, 178–184. [Google Scholar]
- Selvendiran, K.; Bratasz, A.; Kuppusamy, M.L.; Tazi, M.F.; Rivera, B.K.; Kuppusamy, P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int. J. Cancer 2009, 125, 2198–2204. [Google Scholar] [CrossRef]
- Park, S.Y.; Billiar, T.R.; Seol, D.W. Hypoxia inhibition of apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (trail). Biochem. Biophys. Res. Commun. 2002, 291, 150–153. [Google Scholar] [CrossRef]
- Adams, J.M.; Difazio, L.T.; Rolandelli, R.H.; Lujan, J.J.; Hasko, G.; Csoka, B.; Selmeczy, Z.; Nemeth, Z.H. Hif-1: A key mediator in hypoxia. Acta Physiol. Hung. 2009, 96, 19–28. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Treatment of kidney cancer: Insights provided by the vhl tumor-suppressor protein. Cancer 2009, 115, 2262–2272. [Google Scholar] [CrossRef]
- Hu, Y.; Kirito, K.; Yoshida, K.; Mitsumori, T.; Nakajima, K.; Nozaki, Y.; Hamanaka, S.; Nagashima, T.; Kunitama, M.; Sakoe, K.; et al. Inhibition of hypoxia-inducible factor-1 function enhances the sensitivity of multiple myeloma cells to melphalan. Mol. Cancer Ther. 2009, 8, 2329–2338. [Google Scholar]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-alpha binding and ubiquitylation by the von hippel-lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ros but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ros production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef]
- Mansfield, K.D.; Guzy, R.D.; Pan, Y.; Young, R.M.; Cash, T.P.; Schumacker, P.T.; Simon, M.C. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic hif-alpha activation. Cell Metab. 2005, 1, 393–399. [Google Scholar] [CrossRef]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A. Identification and characterization of a new member of the tnf family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Griffith, T.S.; Anderson, R.D.; Davidson, B.L.; Williams, R.D.; Ratliff, T.L. Adenoviral-mediated transfer of the tnf-related apoptosis-inducing ligand/apo-2 ligand gene induces tumor cell apoptosis. J. Immunol. 2000, 165, 2886–2894. [Google Scholar]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis- inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Walsh, S.; Gill, C.; O’Neill, A.; Fitzpatrick, J.M.; Watson, R.W. Hypoxia increases normal prostate epithelial cell resistance to receptor-mediated apoptosis via akt activation. Int. J. Cancer 2009, 124, 1871–1878. [Google Scholar] [CrossRef]
- Kim, M.; Park, S.Y.; Pai, H.S.; Kim, T.H.; Billiar, T.R.; Seol, D.W. Hypoxia inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by blocking bax translocation. Cancer Res. 2004, 64, 4078–4081. [Google Scholar] [CrossRef]
- Ao, J.E.; Kuang, L.H.; Zhou, Y.; Zhao, R.; Yang, C.M. Hypoxia-inducible factor 1 regulated arc expression mediated hypoxia induced inactivation of the intrinsic death pathway in p53 deficient human colon cancer cells. Biochem. Biophys. Res. Commun. 2112, 420, 913–917. [Google Scholar]
- Jeong, J.K.; Moon, M.H.; Seo, J.S.; Seol, J.W.; Lee, Y.J.; Park, S.Y. Sulforaphane blocks hypoxia-mediated resistance to trail-induced tumor cell death. Mol. Med. Rep. 2011, 4, 325–330. [Google Scholar]
- Kim, K.J.; Choi, J.S.; Kang, I.; Kim, K.W.; Jeong, C.H.; Jeong, J.W. Melatonin suppresses tumor progression by reducing angiogenesis stimulated by hif-1 in a mouse tumor model. J. Pineal Res. 2013, 54, 264–270. [Google Scholar] [CrossRef]
- Park, J.W.; Hwang, M.S.; Suh, S.I.; Baek, W.K. Melatonin down-regulates HIF-1α expression through inhibition of protein translation in prostate cancer cells. J. Pineal Res. 2009, 46, 415–421. [Google Scholar] [CrossRef]
- Santoro, R.; Marani, M.; Blandino, G.; Muti, P.; Strano, S. Melatonin triggers p53ser phosphorylation and prevents DNA damage accumulation. Oncogene 2012, 31, 2931–2942. [Google Scholar] [CrossRef]
- Seo, J.S.; Seol, J.W.; Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Park, S.Y. Hypoxia protects neuronal cells from human prion protein fragment-induced apoptosis. J. Neurochem. 2010, 112, 715–722. [Google Scholar] [CrossRef]
- Nagaraj, N.S.; Vigneswaran, N.; Zacharias, W. Hypoxia inhibits trail-induced tumor cell apoptosis: Involvement of lysosomal cathepsins. Apoptosis 2007, 12, 125–139. [Google Scholar] [CrossRef]
- Cho, S.Y.; Lee, H.J.; Jeong, S.J.; Lee, H.J.; Kim, H.S.; Chen, C.Y.; Lee, E.O.; Kim, S.H. Sphingosine kinase 1 pathway is involved in melatonin-induced HIF-1α inactivation in hypoxic pc-3 prostate cancer cells. J. Pineal Res. 2011, 51, 87–83. [Google Scholar] [CrossRef]
- Fujita, N.; Markova, D.; Anderson, D.G.; Chiba, K.; Toyama, Y.; Shapiro, I.M.; Risbud, M.V. Expression of prolyl hydroxylases (phds) is selectively controlled by HIF-1 and HIF-2 proteins in nucleus pulposus cells of the intervertebral disc: Distinct roles of phd2 and phd3 proteins in controlling HIF-1α activity in hypoxia. J. Biol. Chem. 2012, 287, 16975–16986. [Google Scholar] [CrossRef]
- Quast, S.A.; Berger, A.; Plotz, M.; Eberle, J. Sensitization of melanoma cells for trail-induced apoptosis by activation of mitochondrial pathways via bax. Eur. J. Cell Biol. 2014, 93, 42–48. [Google Scholar] [CrossRef]
- Murphy, A.C.; Weyhenmeyer, B.; Noonan, J.; Kilbride, S.M.; Schimansky, S.; Loh, K.P.; Kogel, D.; Letai, A.G.; Prehn, J.H.; Murphy, B.M. Modulation of Mcl-1 sensitizes glioblastoma to trail-induced apoptosis. Apoptosis 2014, 19, 629–642. [Google Scholar]
- Zhu, H.; Guo, W.; Zhang, L.; Davis, J.J.; Wu, S.; Teraishi, F.; Cao, X.; Smythe, W.R.; Fang, B. Enhancing trail-induced apoptosis by Bcl-xL sirna. Cancer Biol. Ther. 2005, 4, 393–397. [Google Scholar] [CrossRef]
- Lamothe, B.; Aggarwal, B.B. Ectopic expression of bcl-2 and bcl-xl inhibits apoptosis induced by tnf-related apoptosis-inducing ligand (trail) through suppression of caspases-8, -7, and -3 and bid cleavage in human acute myelogenous leukemia cell line hl-60. J. Interferon Cytokine Res. 2002, 22, 269–279. [Google Scholar] [CrossRef]
- Chaudhari, A.A.; Seol, J.W.; Kim, S.J.; Lee, Y.J.; Kang, H.S.; Kim, I.S.; Kim, N.S.; Park, S.Y. Reactive oxygen species regulate bax translocation and mitochondrial transmembrane potential, a possible mechanism for enhanced trail-induced apoptosis by cccp. Oncol. Rep. 2007, 18, 71–76. [Google Scholar]
- Park, G.B.; Kim, Y.S.; Lee, H.K.; Song, H.; Kim, S.; Cho, D.H.; Hur, D.Y. Reactive oxygen species and p38 mapk regulate bax translocation and calcium redistribution in salubrinal-induced apoptosis of ebv-transformed b cells. Cancer Lett. 2011, 313, 235–248. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, Y.-J.; Lee, J.-H.; Moon, J.-H.; Park, S.-Y. Overcoming Hypoxic-Resistance of Tumor Cells to TRAIL-Induced Apoptosis through Melatonin. Int. J. Mol. Sci. 2014, 15, 11941-11956. https://doi.org/10.3390/ijms150711941
Lee Y-J, Lee J-H, Moon J-H, Park S-Y. Overcoming Hypoxic-Resistance of Tumor Cells to TRAIL-Induced Apoptosis through Melatonin. International Journal of Molecular Sciences. 2014; 15(7):11941-11956. https://doi.org/10.3390/ijms150711941
Chicago/Turabian StyleLee, You-Jin, Ju-Hee Lee, Ji-Hong Moon, and Sang-Youel Park. 2014. "Overcoming Hypoxic-Resistance of Tumor Cells to TRAIL-Induced Apoptosis through Melatonin" International Journal of Molecular Sciences 15, no. 7: 11941-11956. https://doi.org/10.3390/ijms150711941