Cordycepin Inhibits Lipopolysaccharide (LPS)-Induced Tumor Necrosis Factor (TNF)-α Production via Activating AMP-Activated Protein Kinase (AMPK) Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
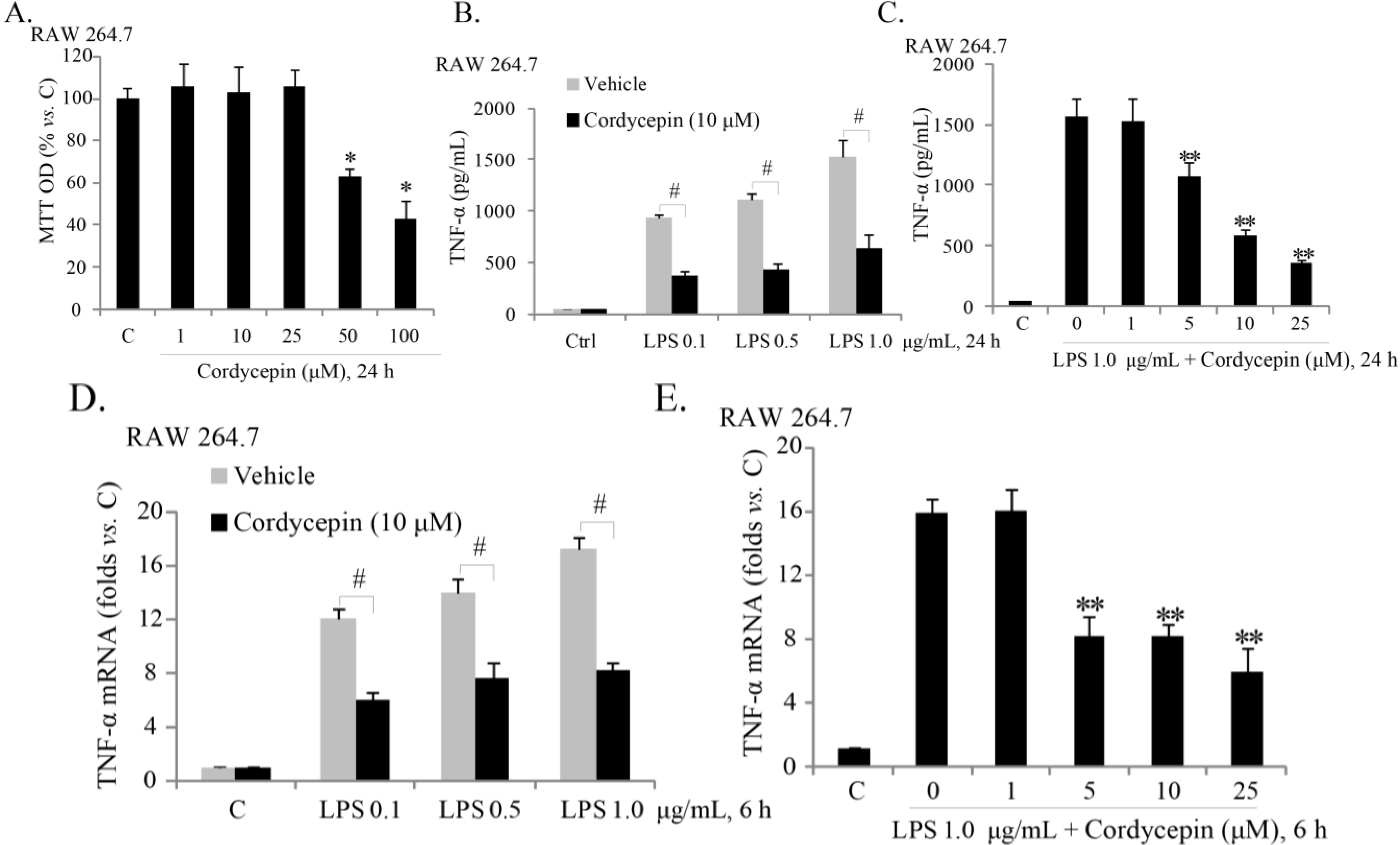
2.1. Sub-Cytotoxic Cordycepin Inhibits LPS (Lipopolysaccharide)-Induced TNFα (Tumor Necrosis Factor α) Production in RAW 264.7 Mouse Macrophages

2.2. Cordycepin Inhibits TNFα Production in ex-Vivo Cultured Peripheral Blood Mononuclear Cells (PBMCs) of KD (Kawasaki Disease) Patients

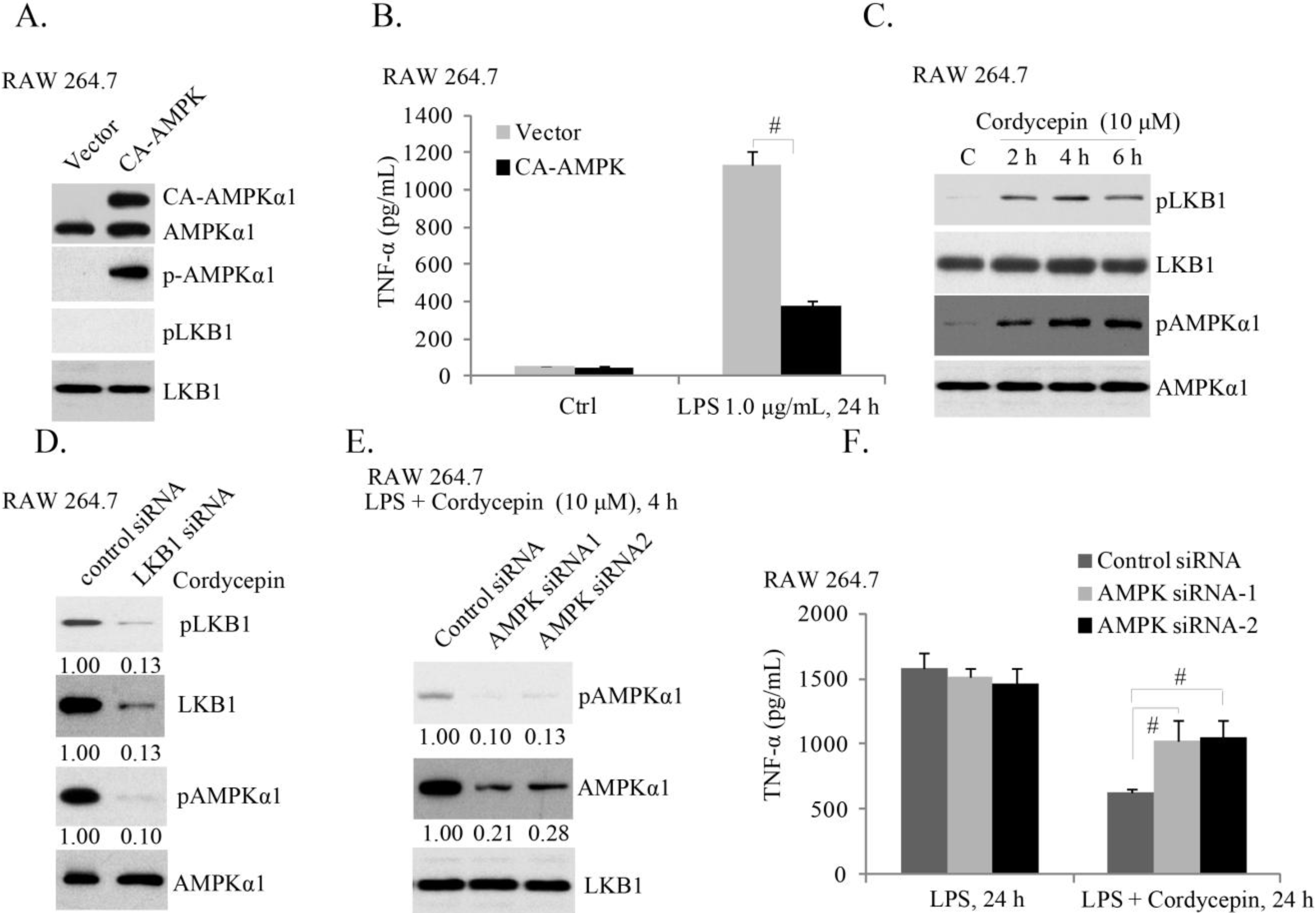
2.3. AMPK (AMP-Activated Protein Kinase) Activation Is Required for Cordycepin’s Effect of LPS-Induced TNFα Production

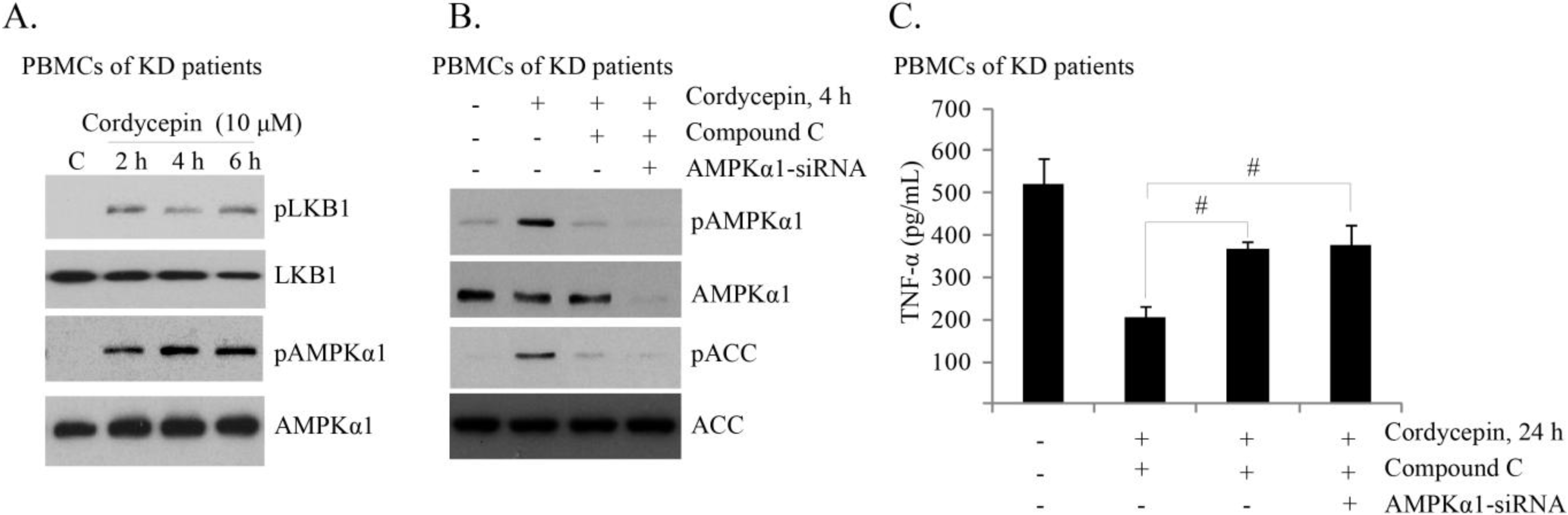
2.4. Cordycepin Activates AMPK to Inhibit TNFα Production in PBMCs of KD Patients

2.5. Cordycepin Inhibits ROS (Reactive Oxygen Species) Production and NF-κB (Nuclear Factor Kappa B) Activation in Both LPS-Stimulated RAW 264.7 Cells and PBMCs from KD Patients

3. Material and Methods
3.1. Chemical and Regents
3.2. Antibodies
3.3. RAW 264.7 Mouse Macrophage Culture
3.4. Bone Marrow–Derived Macrophages (BMDMs) Culture
3.5. Ex Vivo Culture of Peripheral Blood Mononuclear Cells (PBMCs)
3.6. Cell Viability Assay
3.7. TNFα ELISA (Enzyme-Linked Immunosorbent Assay) Assay
3.8. Western-Blots
3.9. Total RNA Isolation and Real-Time Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
3.10. siRNA
3.11. Constitutively Active-AMPK (CA-AMPK) Transfection and Stable Cell Line Selection
3.12. Reactive Oxygen Species (ROS) Assay
3.13. Data Analysis
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dajani, A.S.; Taubert, K.A.; Gerber, M.A.; Shulman, S.T.; Ferrieri, P.; Freed, M.; Takahashi, M.; Bierman, F.Z.; Karchmer, A.W.; Wilson, W.; et al. Diagnosis and therapy of Kawasaki disease in children. Circulation 1993, 87, 1776–1780. [Google Scholar] [CrossRef]
- Kato, H.; Koike, S.; Yokoyama, T. Kawasaki disease: Effect of treatment on coronary artery involvement. Pediatrics 1979, 63, 175–179. [Google Scholar]
- Taubert, K.A.; Rowley, A.H.; Shulman, S.T. Seven-year national survey of Kawasaki disease and acute rheumatic fever. Pediatr. Infect. Dis. J. 1994, 13, 704–708. [Google Scholar] [CrossRef]
- Eberhard, B.A.; Andersson, U.; Laxer, R.M.; Rose, V.; Silverman, E.D. Evaluation of the cytokine response in Kawasaki disease. Pediatr. Infect. Dis. J. 1995, 14, 199–203. [Google Scholar] [CrossRef]
- Lang, B.A.; Silverman, E.D.; Laxer, R.M.; Lau, A.S. Spontaneous tumor necrosis factor production in Kawasaki disease. J. Pediatr. 1989, 115, 939–943. [Google Scholar] [CrossRef]
- Leung, D.Y.; Geha, R.S.; Newburger, J.W.; Burns, J.C.; Fiers, W.; Lapierre, L.A.; Pober, J.S. Two monokines, interleukin 1 and tumor necrosis factor, render cultured vascular endothelial cells susceptible to lysis by antibodies circulating during Kawasaki syndrome. J. Exp. Med. 1986, 164, 1958–1972. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lin, C.C.; Hwang, B.; Chiang, B.N. Cytokines predict coronary aneurysm formation in Kawasaki disease patients. Eur. J. Pediatr. 1993, 152, 309–312. [Google Scholar] [CrossRef]
- Pober, J.S. Endothelial activation: Intracellular signaling pathways. Arthritis Res. 2002, 4, S109–S116. [Google Scholar] [CrossRef]
- Pober, J.S.; Min, W. Endothelial cell dysfunction, injury and death. Handb. Exp. Pharmacol. 2006, 176, 135–156. [Google Scholar] [CrossRef]
- Varani, J.; Ward, P.A. Mechanisms of endothelial cell injury in acute inflammation. Shock 1994, 2, 311–319. [Google Scholar] [CrossRef]
- Andreakos, E.T.; Foxwell, B.M.; Brennan, F.M.; Maini, R.N.; Feldmann, M. Cytokines and anti-cytokine biologicals in autoimmunity: present and future. Cytokine Growth Factor Rev. 2002, 13, 299–313. [Google Scholar] [CrossRef]
- Biedermann, B.C. Vascular endothelium: Checkpoint for inflammation and immunity. News Physiol. Sci. 2001, 16, 84–88. [Google Scholar]
- Hui-Yuen, J.S.; Duong, T.T.; Yeung, R.S. TNF-α is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. J. Immunol. 2006, 176, 6294–6301. [Google Scholar] [CrossRef]
- Son, M.B.; Gauvreau, K.; Burns, J.C.; Corinaldesi, E.; Tremoulet, A.H.; Watson, V.E.; Baker, A.; Fulton, D.R.; Sundel, R.P.; Newburger, J.W. Infliximab for intravenous immunoglobulin resistance in Kawasaki disease: a retrospective study. J. Pediatr. 2011, 158, 644–649. [Google Scholar] [CrossRef]
- Burns, J.C.; Mason, W.H.; Hauger, S.B.; Janai, H.; Bastian, J.F.; Wohrley, J.D.; Balfour, I.; Shen, C.A.; Michel, E.D.; Shulman, S.T.; et al. Infliximab treatment for refractory Kawasaki syndrome. J. Pediatr. 2005, 146, 662–667. [Google Scholar] [CrossRef]
- Oishi, T.; Fujieda, M.; Shiraishi, T.; Ono, M.; Inoue, K.; Takahashi, A.; Ogura, H.; Wakiguchi, H. Infliximab treatment for refractory Kawasaki disease with coronary artery aneurysm. Circ. J. 2008, 72, 850–852. [Google Scholar] [CrossRef]
- Shen, J.; Liang, L.; Wang, C. Perifosine inhibits lipopolysaccharide (LPS)-induced tumor necrosis factor (TNF)-α production via regulation multiple signaling pathways: New implication for Kawasaki disease (KD) treatment. Biochem. Biophys. Res. Commun. 2013, 437, 250–255. [Google Scholar] [CrossRef]
- Shi, P.; Huang, Z.; Tan, X.; Chen, G. Proteomic detection of changes in protein expression induced by cordycepin in human hepatocellular carcinoma BEL-7402 cells. Methods Find. Exp. Clin. Pharmacol. 2008, 30, 347–353. [Google Scholar] [CrossRef]
- Chang, W.; Lim, S.; Song, H.; Song, B.W.; Kim, H.J.; Cha, M.J.; Sung, J.M.; Kim, T.W.; Hwang, K.C. Cordycepin inhibits vascular smooth muscle cell proliferation. Eur. J. Pharmacol. 2008, 597, 64–69. [Google Scholar] [CrossRef]
- Ng, T.B.; Wang, H.X. Pharmacological actions of Cordyceps, a prized folk medicine. J. Pharm. Pharmacol. 2005, 57, 1509–1519. [Google Scholar] [CrossRef]
- Wong, Y.Y.; Moon, A.; Duffin, R.; Barthet-Barateig, A.; Meijer, H.A.; Clemens, M.J.; de Moor, C.H. Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. J. Biol. Chem. 2010, 285, 2610–2621. [Google Scholar]
- Tuli, H.S.; Sharma, A.K.; Sandhu, S.S.; Kashyap, D. Cordycepin: A bioactive metabolite with therapeutic potential. Life Sci. 2013, 93, 863–869. [Google Scholar]
- Zhang, D.W.; Wang, Z.L.; Qi, W.; Lei, W.; Zhao, G.Y. Cordycepin (3'-deoxyadenosine) down-regulates the proinflammatory cytokines in inflammation-induced osteoporosis model. Inflammation 2014. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.; Qu, K.; Zhu, P.; Guo, N.; Zhang, R.; Abliz, Z.; Yu, H.; Zhu, H. Binding of cordycepin monophosphate to AMP-activated protein kinase and its effect on AMP-activated protein kinase activation. Chem. Biol. Drug Des. 2010, 76, 340–344. [Google Scholar] [CrossRef]
- Thomadaki, H.; Scorilas, A.; Tsiapalis, C.M.; Havredaki, M. The role of cordycepin in cancer treatment via induction or inhibition of apoptosis: Implication of polyadenylation in a cell type specific manner. Cancer Chemother. Pharmacol. 2008, 61, 251–265. [Google Scholar]
- Wu, W.C.; Hsiao, J.R.; Lian, Y.Y.; Lin, C.Y.; Huang, B.M. The apoptotic effect of cordycepin on human OEC-M1 oral cancer cell line. Cancer Chemother. Pharmacol 2007, 60, 103–111. [Google Scholar] [CrossRef]
- Tang, J.; Feng, Y.; Tsao, S.; Wang, N.; Curtain, R.; Wang, Y. Berberine and Coptidis rhizoma as novel antineoplastic agents: a review of traditional use and biomedical investigations. J. Ethnopharmacol. 2009, 126, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Ducommun, S.; Ford, R.J.; Bultot, L.; Deak, M.; Bertrand, L.; Kemp, B.E.; Steinberg, G.R.; Sakamoto, K. Enhanced activation of cellular AMPK by dual-small molecule treatment: AICAR and A769662. Am. J. Physiol. Endocrinol. Metab. 2014, 30, 688–696. [Google Scholar]
- Zheng, Q.Y.; Jin, F.S.; Yao, C.; Zhang, T.; Zhang, G.H.; Ai, X. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem. Biophys. Res. Commun. 2012, 419, 741–747. [Google Scholar] [CrossRef]
- Wu, C.; Guo, Y.; Su, Y.; Zhang, X.; Luan, H.; Zhu, H.; He, H.; Wang, X.; Sun, G.; Sun, X.; et al. Cordycepin activates AMP-activated protein kinase (AMPK) via interaction with the gamma1 subunit. J. Cell. Mol. Med. 2014, 18, 293–304. [Google Scholar] [CrossRef]
- Zhao, Y.; Joshi-Barve, S.; Barve, S.; Chen, L.H. Eicosapentaenoic acid prevents LPS-induced TNF-α expression by preventing NF-κB activation. J. Am. Coll. Nutr. 2004, 23, 71–78. [Google Scholar] [CrossRef]
- Chandel, N.S.; Trzyna, W.C.; McClintock, D.S.; Schumacker, P.T. Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin. J. Immunol. 2000, 165, 1013–1021. [Google Scholar] [CrossRef]
- Yuan, H.; Perry, C.N.; Huang, C.; Iwai-Kanai, E.; Carreira, R.S.; Glembotski, C.C.; Gottlieb, R.A. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 470–479. [Google Scholar]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Girish, M.; Subramaniam, G. Infliximab treatment in refractory Kawasaki syndrome. Indian J. Pediatr. 2008, 75, 521–522. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell. Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Zhao, X.; Zmijewski, J.W.; Lorne, E.; Liu, G.; Park, Y.J.; Tsuruta, Y.; Abraham, E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, 497–504. [Google Scholar] [CrossRef]
- Cacicedo, J.M.; Yagihashi, N.; Keaney, J.F., Jr.; Ruderman, N.B.; Ido, Y. AMPK inhibits fatty acid-induced increases in NF-κB transactivation in cultured human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 324, 1204–1209. [Google Scholar] [CrossRef]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-κB. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef]
- Sanlioglu, S.; Williams, C.M.; Samavati, L.; Butler, N.S.; Wang, G.; McCray, P.B., Jr.; Ritchie, T.C.; Hunninghake, G.W.; Zandi, E.; Engelhardt, J.F. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-κB. J. Biol. Chem. 2001, 276, 30188–30198. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, J.-L.; Xu, Y.; Shen, J. Cordycepin Inhibits Lipopolysaccharide (LPS)-Induced Tumor Necrosis Factor (TNF)-α Production via Activating AMP-Activated Protein Kinase (AMPK) Signaling. Int. J. Mol. Sci. 2014, 15, 12119-12134. https://doi.org/10.3390/ijms150712119
Zhang J-L, Xu Y, Shen J. Cordycepin Inhibits Lipopolysaccharide (LPS)-Induced Tumor Necrosis Factor (TNF)-α Production via Activating AMP-Activated Protein Kinase (AMPK) Signaling. International Journal of Molecular Sciences. 2014; 15(7):12119-12134. https://doi.org/10.3390/ijms150712119
Chicago/Turabian StyleZhang, Jian-Li, Ying Xu, and Jie Shen. 2014. "Cordycepin Inhibits Lipopolysaccharide (LPS)-Induced Tumor Necrosis Factor (TNF)-α Production via Activating AMP-Activated Protein Kinase (AMPK) Signaling" International Journal of Molecular Sciences 15, no. 7: 12119-12134. https://doi.org/10.3390/ijms150712119