Making the Bend: DNA Tertiary Structure and Protein-DNA Interactions

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
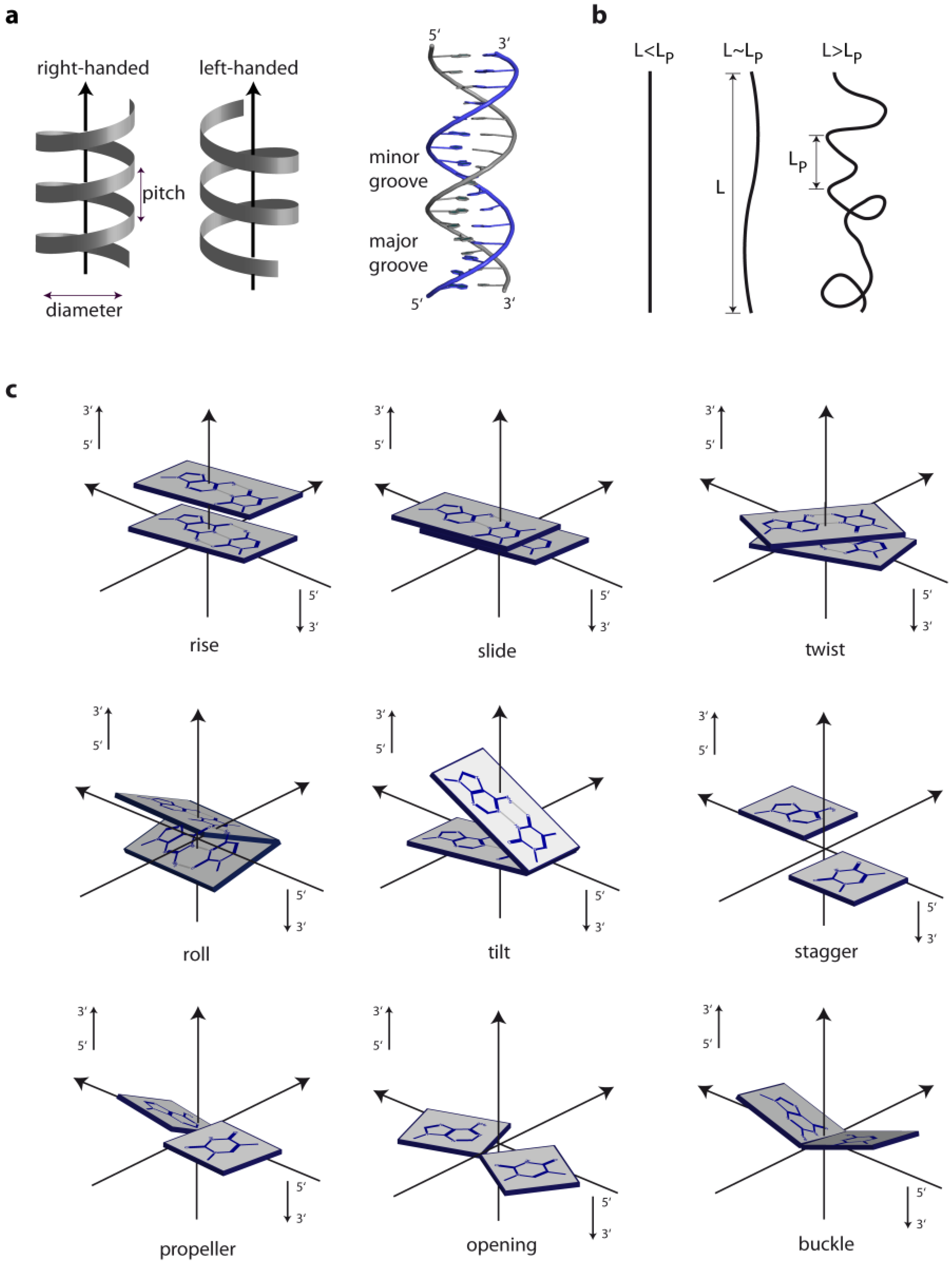
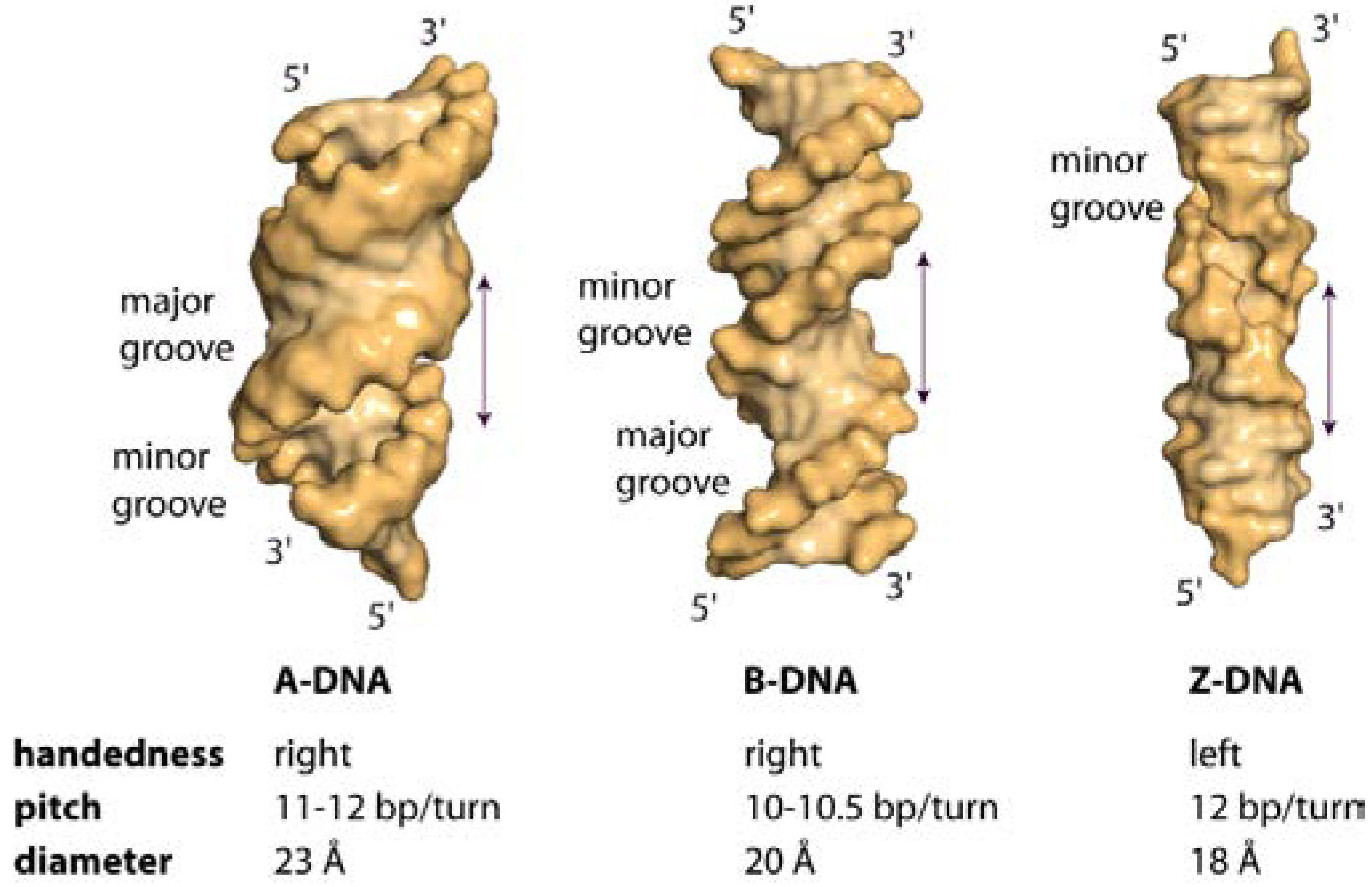
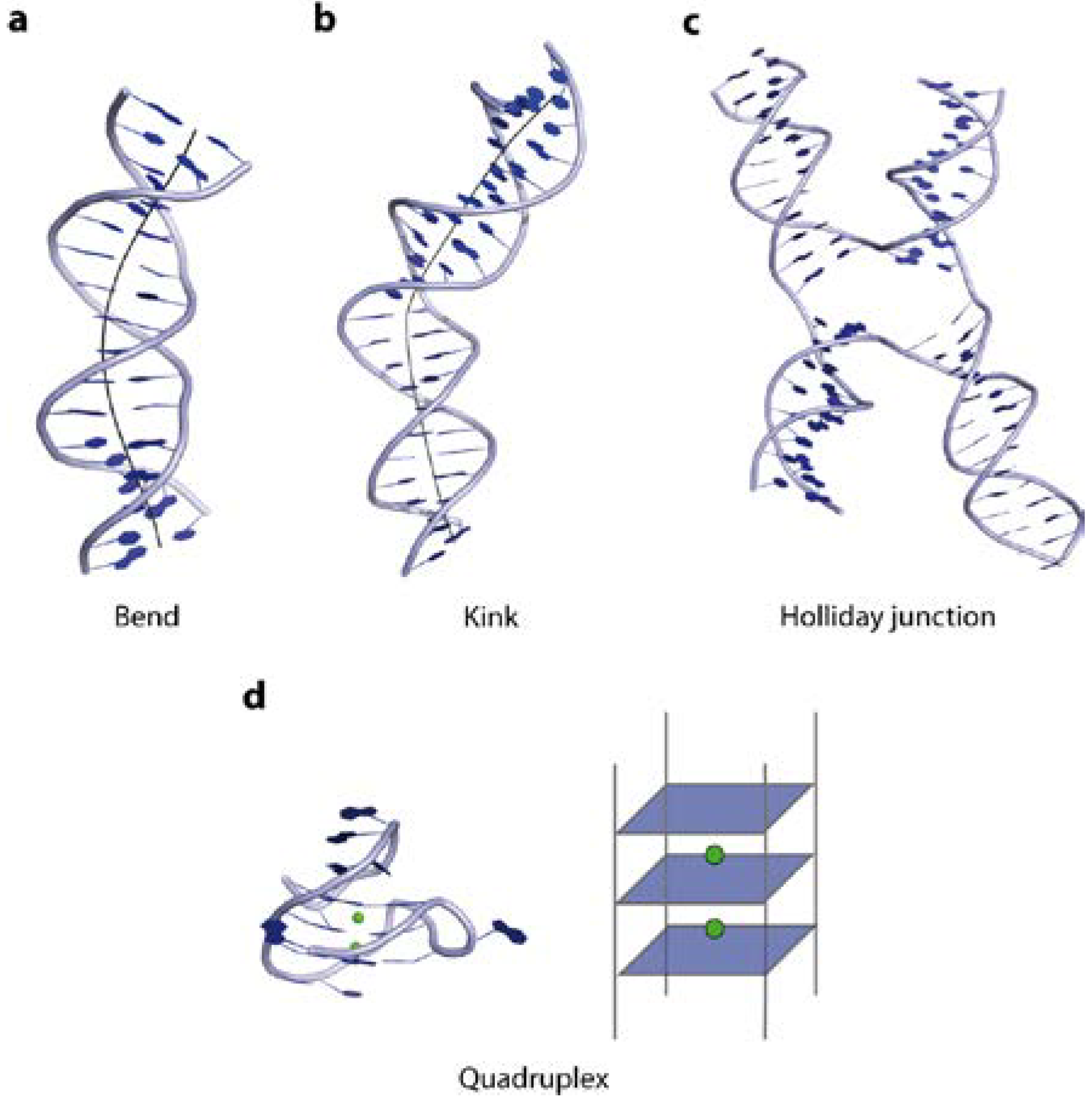
1. The Overall Topology of DNA


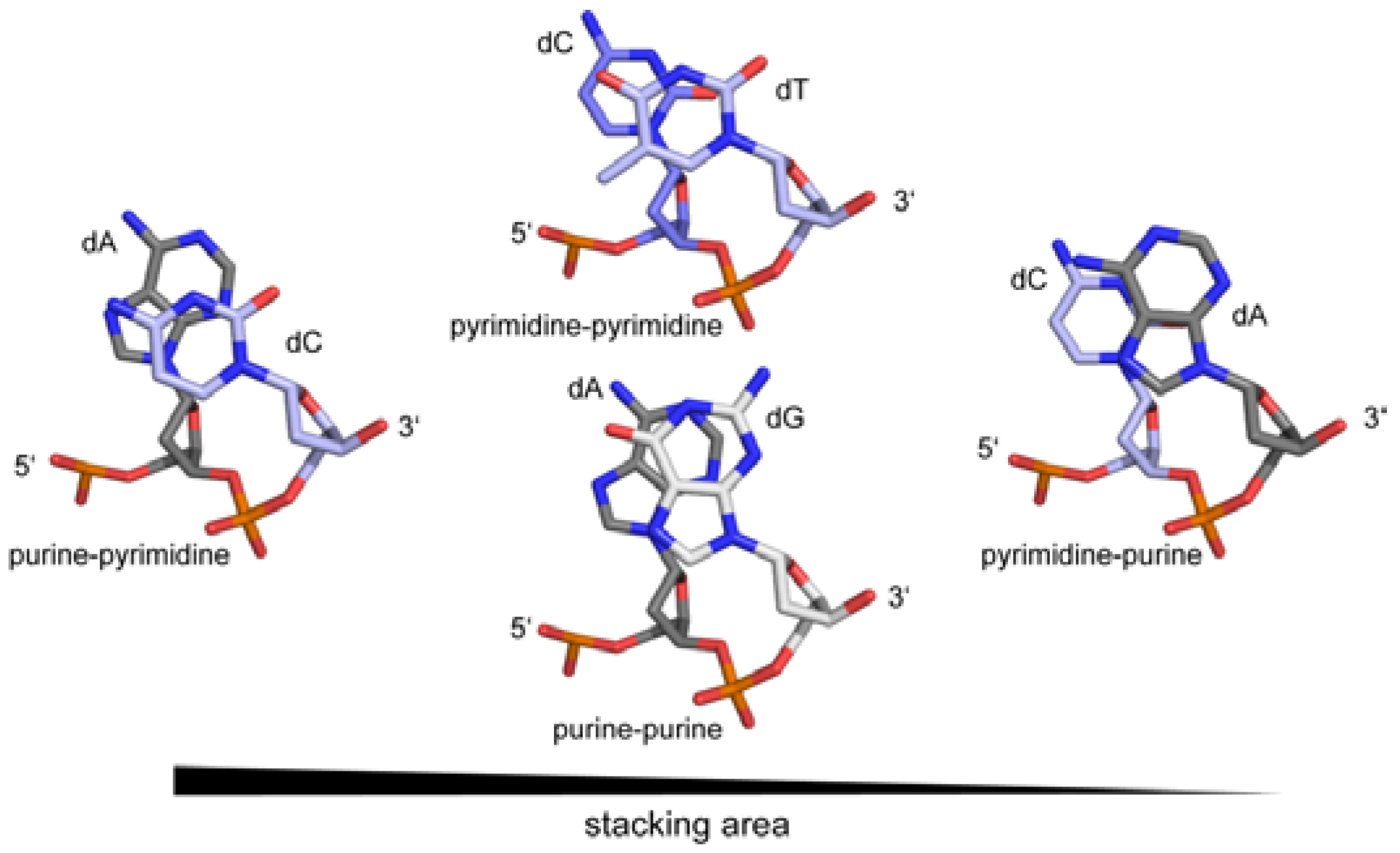
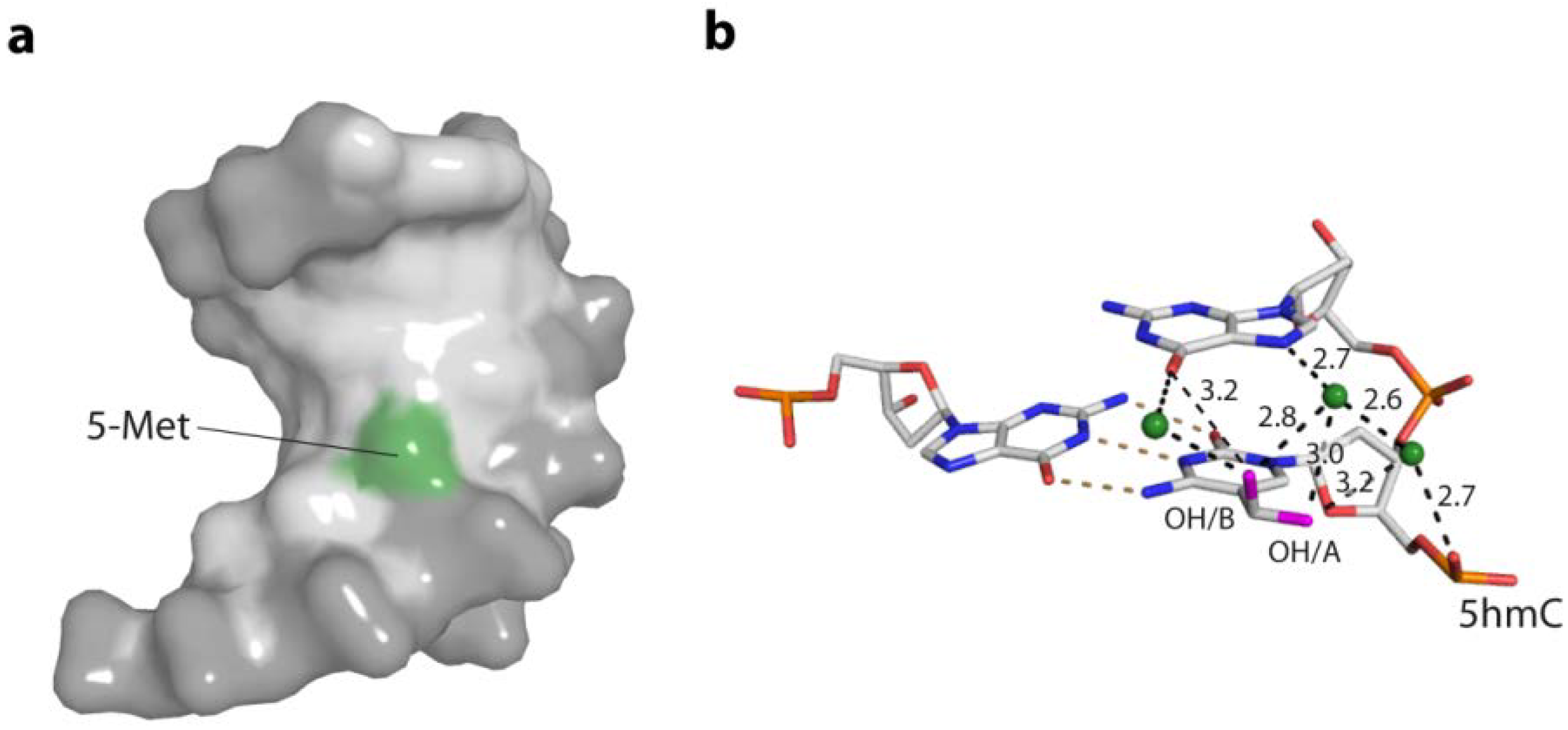
2. DNA Sequence and DNA Structure




3. Thermodynamic Consideration of DNA-Protein Interactions
4. Base Readout, Pre-Shaped DNA and Protein Recognition

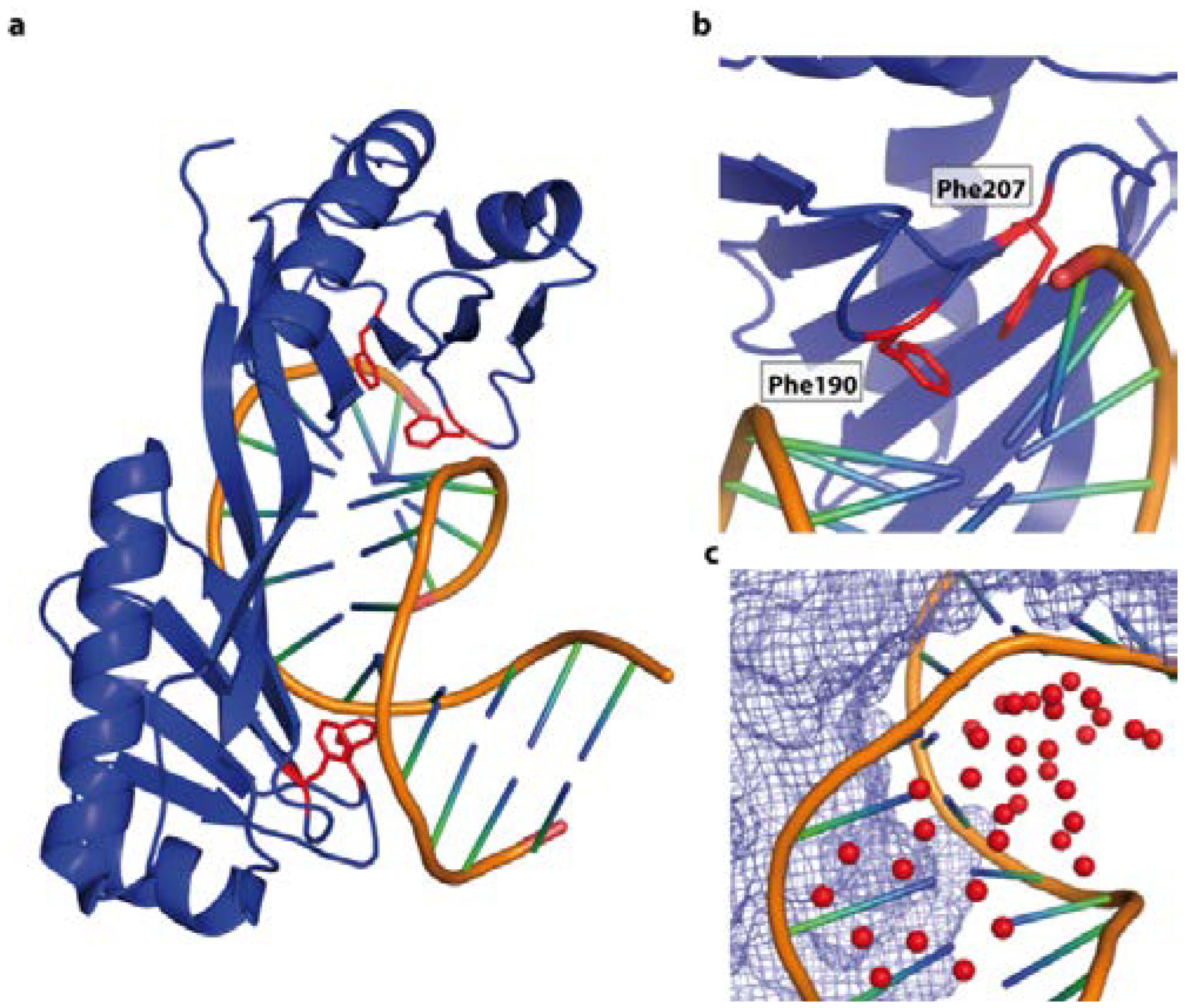
4.1. Base Readout and Recognition Sequence Intrinsic Shape Readout—The Restriction Endonucleases HindIII and EcoRV

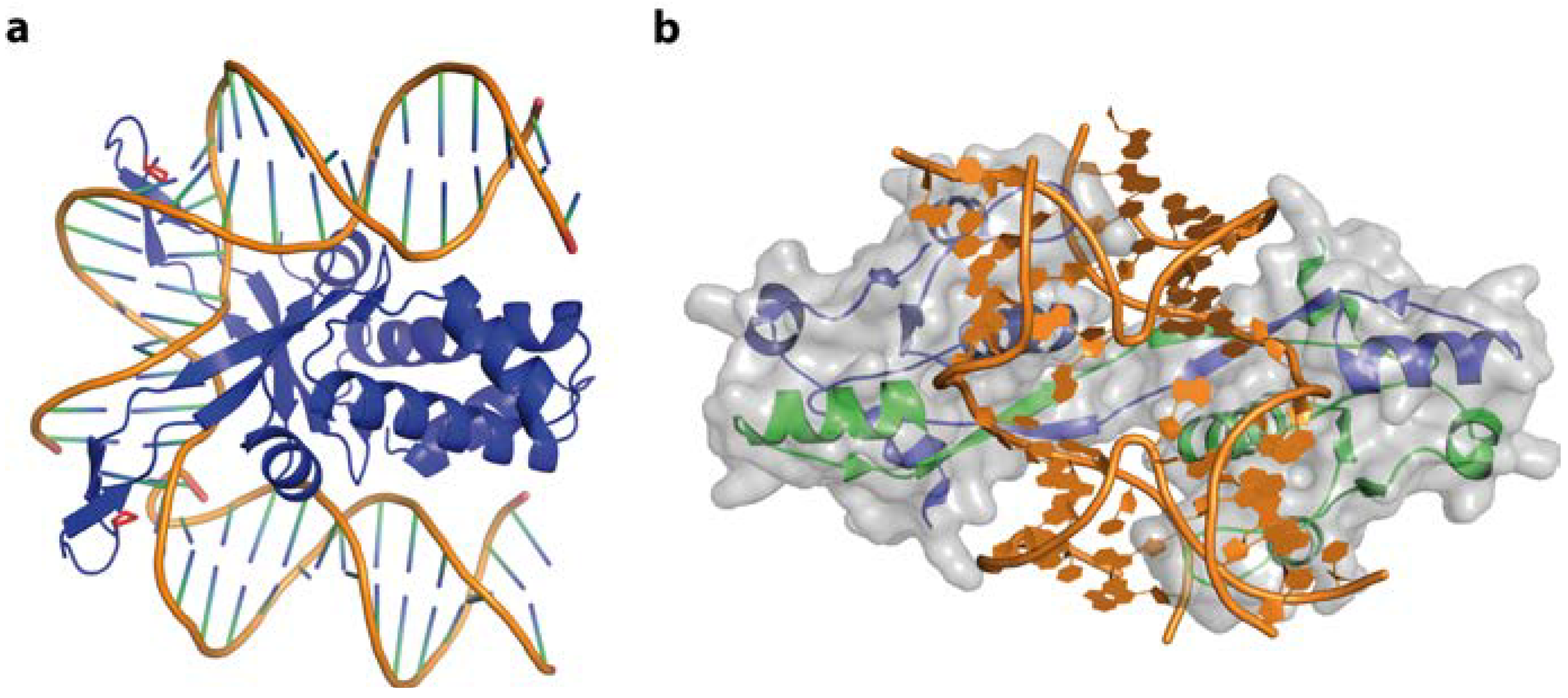
4.2. Combining Base and Shape Readout—The Escherichia coli Trp Operator

4.3. Base Readout and DNA Shape Context—The Escherichia coli LexA Repressor

4.4. Sequence Specific Shape Readout—The TATA Binding Protein and the TATA Box

4.5. DNA Shape Recognition
4.5.1. The Bacterial Chromosomal Proteins HU and IHF

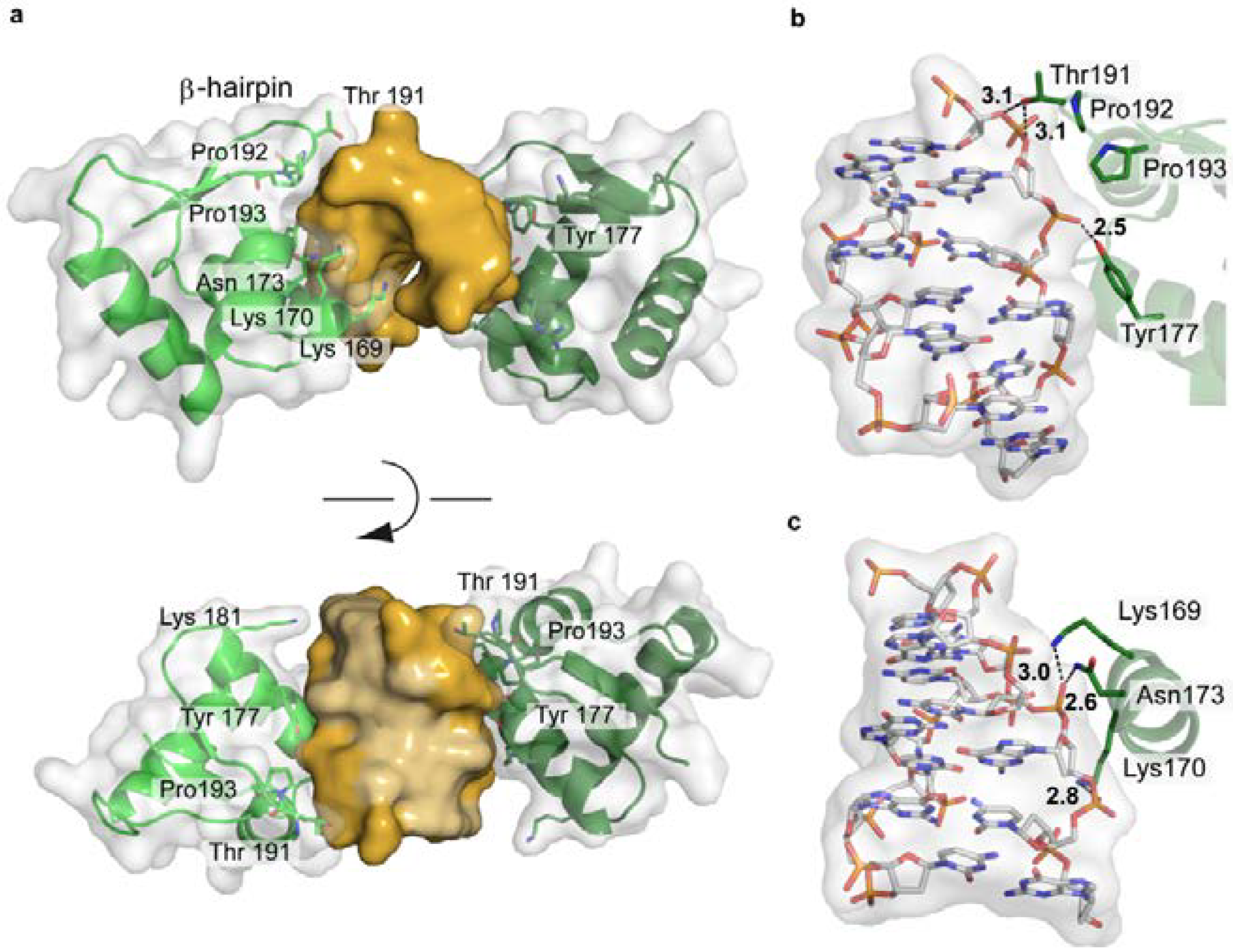
4.5.2. The Holliday Junction and T7 Endonuclease I
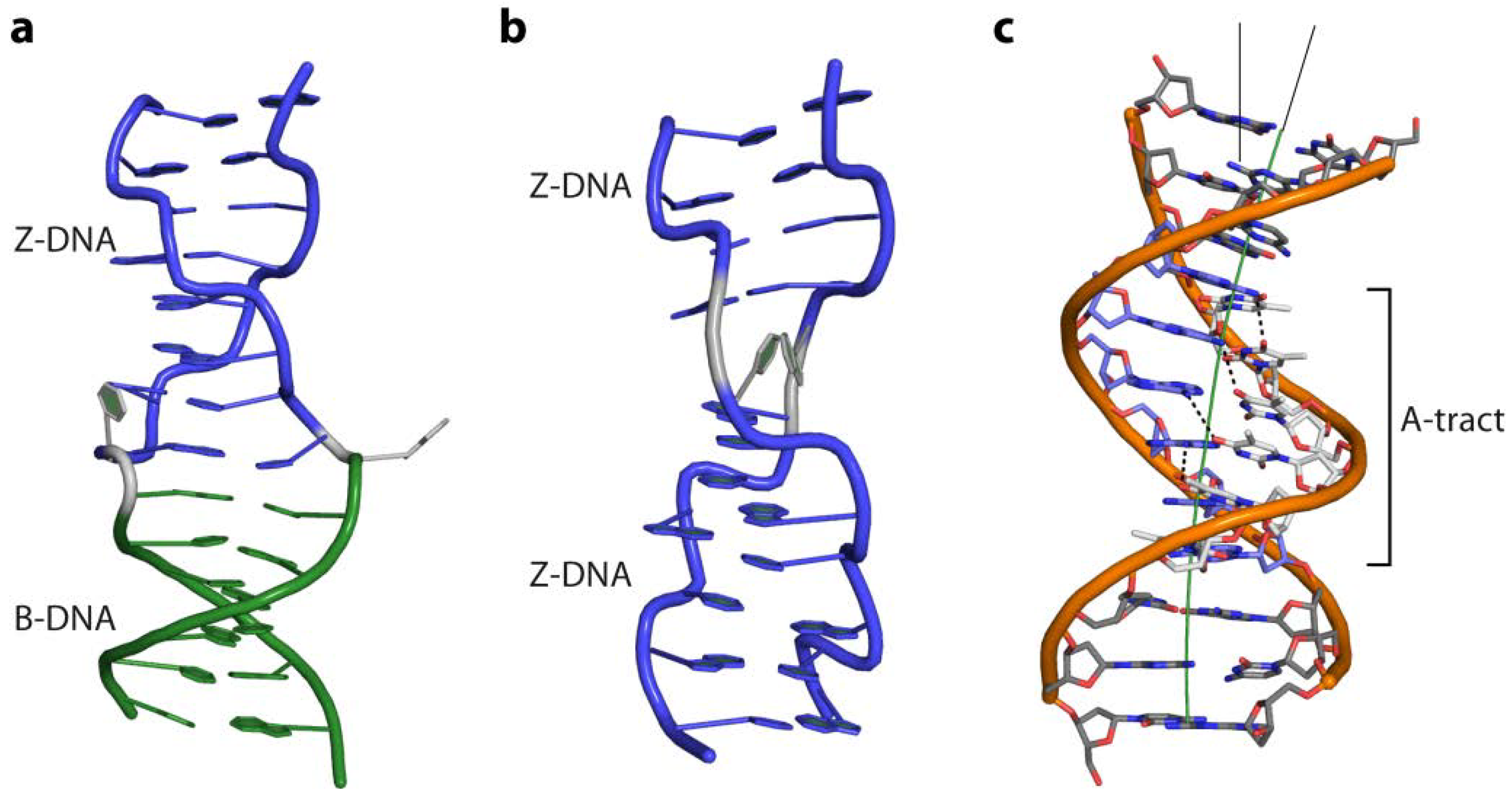
4.5.3. Z-DNA and ADAR1

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dickerson, R.E. DNA structure from A to Z. Methods Enzymol. 1992, 211, 67–111. [Google Scholar] [CrossRef]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark, A.D.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark, P.; et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6320–6324. [Google Scholar] [CrossRef]
- Lu, X.J.; Shakked, Z.; Olson, W.K. A-form conformational motifs in ligand-bound DNA structures. J. Mol. Biol. 2000, 300, 819–840. [Google Scholar] [CrossRef]
- Petersen, M.; Bondensgaard, K.; Wengel, J.; Jacobsen, J.P. Locked nucleic acid (LNA) recognition of RNA: NMR solution structures of LNA:RNA hybrids. J. Am. Chem. Soc. 2002, 124, 5974–5982. [Google Scholar] [CrossRef]
- Ban, C.; Ramakrishnan, B.; Sundaralingam, M. A single 2'-hydroxyl group converts B-DNA to A-DNA: Crystal structure of the DNA-RNA chimeric decamer duplex d(CCGGC)r(G)d(CCGG) with a novel intermolecular G C base-paired quadruplet. J. Mol. Biol. 1994, 236, 275–285. [Google Scholar] [CrossRef]
- Chen, X.; Ramakrishnan, B.; Sundaralingam, M. Crystal structures of B-form DNA-RNA chimers complexed with distamycin. Nat. Struct. Biol. 1995, 2, 733–735. [Google Scholar] [CrossRef]
- Wang, A.H.; Quigley, G.J.; Kolpak, F.J.; Crawford, J.L.; van Boom, J.H.; van der Marel, G.; Rich, A. Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature 1979, 282, 680–686. [Google Scholar] [CrossRef]
- Rich, A.; Zhang, S. Timeline: Z-DNA: The long road to biological function. Nat. Rev. Genet. 2003, 4, 566–572. [Google Scholar] [CrossRef]
- Dickerson, R.E. Definitions and nomenclature of nucleic acid structure components. Nucleic Acids Res. 1989, 17, 1797–1803. [Google Scholar]
- Olson, W.K.; Bansal, M.; Burley, S.K.; Dickerson, R.E.; Gerstein, M.; Harvey, S.C.; Heinemann, U.; Lu, X.J.; Neidle, S.; Shakked, Z.; et al. A standard reference frame for the description of nucleic acid base-pair geometry. J. Mol. Biol. 2001, 313, 229–237. [Google Scholar] [CrossRef]
- Sinden, R.R. DNA Structure and Function; Academic Press: New York, NY, USA, 1994. [Google Scholar]
- Rao, A.N.; Grainger, D.W. Biophysical properties of nucleic acids at surfaces relevant to microarray performance. Biomater. Sci. 2014, 2, 436–471. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. 3DNA: A versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008, 3, 1213–1227. [Google Scholar] [CrossRef]
- Kitayner, M.; Rozenberg, H.; Rohs, R.; Suad, O.; Rabinovich, D.; Honig, B.; Shakked, Z. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010, 17, 423–429. [Google Scholar] [CrossRef]
- Rohs, R.; West, S.M.; Sosinsky, A.; Liu, P.; Mann, R.S.; Honig, B. The role of DNA shape in protein-DNA recognition. Nature 2009, 461, 1248–1253. [Google Scholar] [CrossRef]
- Parker, S.C.; Hansen, L.; Abaan, H.O.; Tullius, T.D.; Margulies, E.H. Local DNA topography correlates with functional noncoding regions of the human genome. Science 2009, 324, 389–392. [Google Scholar] [CrossRef]
- Vafabakhsh, R.; Ha, T. Extreme bendability of DNA less than 100 base pairs long revealed by single-molecule cyclization. Science 2012, 337, 1097–1101. [Google Scholar] [CrossRef]
- Becker, N.A.; Kahn, J.D.; Maher, L.J. Bacterial repression loops require enhanced DNA flexibility. J. Mol. Biol. 2005, 349, 716–730. [Google Scholar] [CrossRef]
- Virstedt, J.; Berge, T.; Henderson, R.M.; Waring, M.J.; Travers, A.A. The influence of DNA stiffness upon nucleosome formation. J. Struct. Biol. 2004, 148, 66–85. [Google Scholar] [CrossRef]
- Buttinelli, M.; Minnock, A.; Panetta, G.; Waring, M.; Travers, A. The exocyclic groups of DNA modulate the affinity and positioning of the histone octamer. Proc. Natl. Acad. Sci. USA 1998, 95, 8544–8549. [Google Scholar]
- Diekmann, S.; von Kitzing, E.; McLaughlin, L.; Ott, J.; Eckstein, F. The influence of exocyclic substituents of purine bases on DNA curvature. Proc. Natl. Acad. Sci. USA 1987, 84, 8257–8261. [Google Scholar] [CrossRef]
- Nelson, H.C.; Finch, J.T.; Luisi, B.F.; Klug, A. The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature 1987, 330, 221–226. [Google Scholar] [CrossRef]
- Protozanova, E.; Yakovchuk, P.; Frank-Kamenetskii, M.D. Stacked-unstacked equilibrium at the nick site of DNA. J. Mol. Biol. 2004, 342, 775–785. [Google Scholar] [CrossRef]
- Yakovchuk, P.; Protozanova, E.; Frank-Kamenetskii, M.D. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006, 34, 564–574. [Google Scholar] [CrossRef]
- Burkhoff, A.M.; Tullius, T.D. Structural details of an adenine tract that does not cause DNA to bend. Nature 1988, 331, 455–457. [Google Scholar] [CrossRef]
- Haran, T.E.; Mohanty, U. The unique structure of A-tracts and intrinsic DNA bending. Q. Rev. Biophys. 2009, 42, 41–81. [Google Scholar] [CrossRef]
- Wiggins, P.A.; van der Heijden, T.; Moreno-Herrero, F.; Spakowitz, A.; Phillips, R.; Widom, J.; Dekker, C.; Nelson, P.C. High flexibility of DNA on short length scales probed by atomic force microscopy. Nat. Nanotechnol. 2006, 1, 137–141. [Google Scholar] [CrossRef]
- Savelyev, A.; Materese, C.K.; Papoian, G.A. Is DNA’s rigidity dominated by electrostatic or nonelectrostatic interactions? J. Am. Chem. Soc. 2011, 133, 19290–19293. [Google Scholar] [CrossRef]
- Svozil, D.; Kalina, J.; Omelka, M.; Schneider, B. DNA conformations and their sequence preferences. Nucleic Acids Res. 2008, 36, 3690–3706. [Google Scholar] [CrossRef]
- Johnston, B.H.; Quigley, G.J.; Ellison, M.J.; Rich, A. The Z-Z junction: The boundary between two out-of-phase Z-DNA regions. Biochemistry 1991, 30, 5257–5263. [Google Scholar] [CrossRef]
- Wang, G.; Christensen, L.A.; Vasquez, K.M. Z-DNA-forming sequences generate large-scale deletions in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2677–2682. [Google Scholar] [CrossRef]
- Liu, R.; Liu, H.; Chen, X.; Kirby, M.; Brown, P.O.; Zhao, K.J. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell 2001, 106, 309–318. [Google Scholar] [CrossRef]
- Oh, D.B.; Kim, Y.G.; Rich, A. Z-DNA-binding proteins can act as potent effectors of gene expression in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 16666–16671. [Google Scholar]
- Garner, M.M.; Felsenfeld, G. Effect of Z-DNA on nucleosome placement. J. Mol. Biol. 1987, 196, 581–590. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef]
- Herbert, A.; Lowenhaupt, K.; Spitzner, J.; Rich, A. Chicken double-stranded RNA adenosine deaminase has apparent specificity for Z-DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 7550–7554. [Google Scholar] [CrossRef]
- Kim, Y.G.; Muralinath, M.; Brandt, T.; Pearcy, M.; Hauns, K.; Lowenhaupt, K.; Jacobs, B.L.; Rich, A. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 6974–6979. [Google Scholar]
- Rothenburg, S.; Deigendesch, N.; Dittmar, K.; Koch-Nolte, F.; Haag, F.; Lowenhaupt, K.; Rich, A. A PKR-like eukaryotic initiation factor 2alpha kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. Proc. Natl. Acad. Sci. USA 2005, 102, 1602–1607. [Google Scholar] [CrossRef]
- Schwartz, T.; Behlke, J.; Lowenhaupt, K.; Heinemann, U.; Rich, A. Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat. Struct. Biol. 2001, 8, 761–765. [Google Scholar] [CrossRef]
- Goodbourn, S.; Didcock, L.; Randall, R.E. Interferons: Cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 2000, 81, 2341–2364. [Google Scholar]
- Peck, L.J.; Wang, J.C. Sequence dependence of the helical repeat of DNA in solution. Nature 1981, 292, 375–378. [Google Scholar] [CrossRef]
- Rhodes, D.; Klug, A. Sequence-dependent helical periodicity of DNA. Nature 1981, 292, 378–380. [Google Scholar] [CrossRef]
- Lavery, R.; Moakher, M.; Maddocks, J.H.; Petkeviciute, D.; Zakrzewska, K. Conformational analysis of nucleic acids revisited: Curves+. Nucleic Acids Res. 2009, 37, 5917–5929. [Google Scholar]
- Struhl, K. Naturally occurring poly(dA-dT) sequences are upstream promoter elements for constitutive transcription in yeast. Proc. Natl. Acad. Sci. USA 1985, 82, 8419–8423. [Google Scholar] [CrossRef]
- Kaplan, N.; Moore, I.K.; Fondufe-Mittendorf, Y.; Gossett, A.J.; Tillo, D.; Field, Y.; LeProust, E.M.; Hughes, T.R.; Lieb, J.D.; Widom, J.; et al. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 2009, 458, 362–366. [Google Scholar] [CrossRef]
- Dickerson, R.E. DNA bending: The prevalence of kinkiness and the virtues of normality. Nucleic Acids Res. 1998, 26, 1906–1926. [Google Scholar] [CrossRef]
- Olson, W.K.; Gorin, A.A.; Lu, X.J.; Hock, L.M.; Zhurkin, V.B. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 11163–11168. [Google Scholar]
- Mack, D.R.; Chiu, T.K.; Dickerson, R.E. Intrinsic bending and deformability at the T-A step of CCTTTAAAGG: A comparative analysis of T-A and A-T steps within A-tracts. J. Mol. Biol. 2001, 312, 1037–1049. [Google Scholar] [CrossRef]
- Rauch, C.; Trieb, M.; Wellenzohn, B.; Loferer, M.; Voegele, A.; Wibowo, F.R.; Liedl, K.R. C5-methylation of cytosine in B-DNA thermodynamically and kinetically stabilizes BI. J. Am. Chem. Soc. 2003, 125, 14990–14991. [Google Scholar] [CrossRef]
- Acosta-Silva, C.; Branchadell, V.; Bertran, J.; Oliva, A. Mutual relationship between stacking and hydrogen bonding in DNA. Theoretical study of guanine-cytosine, guanine-5-methylcytosine, and their dimers. J. Phys. Chem. B 2010, 114, 10217–10227. [Google Scholar]
- Norberg, J.V.; Vihinen, M. Molecular dynamics simulation of the effects of cytosine methylation on structure of oligonucleotides. J. Mol. Struct.Theochem. 2001, 546, 51–62. [Google Scholar] [CrossRef]
- Wanunu, M.; Cohen-Karni, D.; Johnson, R.R.; Fields, L.; Benner, J.; Peterman, N.; Zheng, Y.; Klein, M.L.; Drndic, M. Discrimination of methylcytosine from hydroxymethylcytosine in DNA molecules. J. Am. Chem. Soc. 2011, 133, 486–492. [Google Scholar] [CrossRef]
- Hodges-Garcia, Y.; Hagerman, P.J. Cytosine methylation can induce local distortions in the structure of duplex DNA. Biochemistry 1992, 31, 7595–7599. [Google Scholar] [CrossRef]
- Zou, X.; Ma, W.; Solov’yov, I.A.; Chipot, C.; Schulten, K. Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res. 2012, 40, 2747–2758. [Google Scholar] [CrossRef]
- Davey, C.S.; Pennings, S.; Reilly, C.; Meehan, R.R.; Allan, J. A determining influence for CpG dinucleotides on nucleosome positioning in vitro. Nucleic Acids Res. 2004, 32, 4322–4331. [Google Scholar]
- Jimenez-Useche, I.; Shim, D.; Yu, J.; Yuan, C. Unmethylated and methylated CpG dinucleotides distinctively regulate the physical properties of DNA. Biopolymers 2014, 101, 517–524. [Google Scholar] [CrossRef]
- Lercher, L.; McDonough, M.A.; El-Sagheer, A.H.; Thalhammer, A.; Kriaucionis, S.; Brown, T.; Schofield, C.J. Structural insights into how 5-hydroxymethylation influences transcription factor binding. Chem. Commun. 2014, 50, 1794–1796. [Google Scholar]
- Behe, M.; Felsenfeld, G. Effects of methylation on a synthetic polynucleotide: The B-to-Z transition in poly(dG-m5dC).poly(dG-m5dC). Proc. Natl. Acad. Sci. USA 1981, 78, 1619–1623. [Google Scholar] [CrossRef]
- Duckett, D.R.; Murchie, A.I.; Diekmann, S.; von Kitzing, E.; Kemper, B.; Lilley, D.M.J. The structure of the Holliday junction, and its resolution. Cell 1988, 55, 79–89. [Google Scholar] [CrossRef]
- Liu, Y.; West, S.C. Happy Hollidays: 40th anniversary of the Holliday junction. Nat. Rev. Mol. Cell. Biol. 2004, 5, 937–944. [Google Scholar] [CrossRef]
- Watson, J.; Hays, F.A.; Ho, P.S. Definitions and analysis of DNA Holliday junction geometry. Nucleic Acids Res. 2004, 32, 3017–3027. [Google Scholar] [CrossRef]
- Mirkin, S.M. Discovery of alternative DNA structures: A heroic decade (1979–1989). Front. Biosci. 2008, 13, 1064–1071. [Google Scholar]
- Hyeon, C.; Lee, J.; Yoon, J.; Hohng, S.; Thirumalai, D. Hidden complexity in the isomerization dynamics of Holliday junctions. Nat. Chem. 2012, 4, 907–914. [Google Scholar]
- McKinney, S.A.; Declais, A.C.; Lilley, D.M.; Ha, T. Structural dynamics of individual Holliday junctions. Nat. Struct. Biol. 2003, 10, 93–97. [Google Scholar]
- Gellert, M.; Lipsett, M.N.; Davies, D.R. Helix formation by guanylic acid. Proc. Natl. Acad. Sci. USA 1962, 48, 2013–2018. [Google Scholar] [CrossRef]
- Lam, E.Y.; Beraldi, D.; Tannahill, D.; Balasubramanian, S. G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 2013, 4, 1796. [Google Scholar] [CrossRef]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef]
- Todd, A.K.; Neidle, S. Mapping the sequences of potential guanine quadruplex motifs. Nucleic Acids Res. 2011, 39, 4917–4927. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Klug, A. Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 1989, 342, 825–829. [Google Scholar] [CrossRef]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar]
- Koole, W.; van Schendel, R.; Karambelas, A.E.; van Heteren, J.T.; Okihara, K.L.; Tijsterman, M.A. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun. 2014, 5, 3216. [Google Scholar]
- Zhao, J.; Bacolla, A.; Wang, G.; Vasquez, K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010, 67, 43–62. [Google Scholar] [CrossRef]
- London, T.B.; Barber, L.J.; Mosedale, G.; Kelly, G.P.; Balasubramanian, S.; Hickson, I.D.; Boulton, S.J.; Hiom, K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J. Biol. Chem. 2008, 283, 36132–36139. [Google Scholar] [CrossRef]
- Popuri, V.; Bachrati, C.Z.; Muzzolini, L.; Mosedale, G.; Costantini, S.; Giacomini, E.; Hickson, I.D.; Vindigni, A. The Human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J. Biol. Chem. 2008, 283, 17766–17776. [Google Scholar]
- Eddy, S.; Ketkar, A.; Zafar, M.K.; Maddukuri, L.; Choi, J.Y.; Eoff, R.L. Human Rev1 polymerase disrupts G-quadruplex DNA. Nucleic Acids Res. 2014, 42, 3272–3285. [Google Scholar]
- Lopes, J.; Piazza, A.; Bermejo, R.; Kriegsman, B.; Colosio, A.; Teulade-Fichou, M.P.; Foiani, M.; Nicolas, A. G-quadruplex-induced instability during leading-strand replication. EMBO J. 2011, 30, 4033–4046. [Google Scholar] [CrossRef]
- Ribeyre, C.; Lopes, J.; Boule, J.B.; Piazza, A.; Guedin, A.; Zakian, V.A.; Mergny, J.L.; Nicolas, A. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 2009, 5, e1000475. [Google Scholar]
- Jen-Jacobson, L. Protein-DNA recognition complexes: Conservation of structure and binding energy in the transition state. Biopolymers 1997, 44, 153–180. [Google Scholar] [CrossRef]
- Lin, S.; Riggs, A.D. A comparison of lac repressor binding to operator and to nonoperator DNA. Biochem. Biophys. Res. Commun. 1975, 62, 704–710. [Google Scholar] [CrossRef]
- von Hippel, P.H.; Berg, O.G. On the specificity of DNA-protein interactions. Proc. Natl. Acad. Sci. USA 1986, 83, 1608–1612. [Google Scholar] [CrossRef]
- Härd, T.; Lundbäck, T. Thermodynamics of sequence-specific protein-DNA interactions. Biophys. Chem. 1996, 62, 121–139. [Google Scholar] [CrossRef]
- Jen-Jacobson, L.; Engler, L.E.; Jacobson, L.A. Structural and thermodynamic strategies for site-specific DNA binding proteins. Structure 2000, 8, 1015–1023. [Google Scholar] [CrossRef]
- Travers, A.A.; Vaillant, C.; Arneodo, A.; Muskhelishvili, G. DNA structure, nucleosome placement and chromatin remodelling: A perspective. Biochem. Soc. Trans. 2012, 40, 335–340. [Google Scholar]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef]
- Lavery, R. Recognizing DNA. Q. Rev. Biophys. 2005, 38, 339–344. [Google Scholar] [CrossRef]
- Seeman, N.C.; Rosenberg, J.M.; Rich, A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc. Natl. Acad. Sci. USA 1976, 73, 804–808. [Google Scholar] [CrossRef]
- Coulocheri, S.A.; Pigis, D.G.; Papavassiliou, K.A.; Papavassiliou, A.G. Hydrogen bonds in protein-DNA complexes: Where geometry meets plasticity. Biochimie 2007, 89, 1291–1303. [Google Scholar] [CrossRef]
- Gordan, R.; Shen, N.; Dror, I.; Zhou, T.; Horton, J.; Rohs, R.; Bulyk, M.L. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell. Rep. 2013, 3, 1093–1104. [Google Scholar] [CrossRef]
- Harris, L.A.; Watkins, D.; Williams, L.D.; Koudelka, G.B. Indirect readout of DNA sequence by p22 repressor: Roles of DNA and protein functional groups in modulating DNA conformation. J. Mol. Biol. 2013, 425, 133–143. [Google Scholar] [CrossRef]
- Shakked, Z.; Guzikevich-Guerstein, G.; Frolow, F.; Rabinovich, D.; Joachimiak, A.; Sigler, P.B. Determinants of repressor/operator recognition from the structure of the trp operator binding site. Nature 1994, 368, 469–473. [Google Scholar] [CrossRef]
- Romanuka, J.; Folkers, G.E.; Biris, N.; Tishchenko, E.; Wienk, H.; Bonvin, A.M.; Kaptein, R.; Boelens, R. Specificity and affinity of Lac repressor for the auxiliary operators O2 and O3 are explained by the structures of their protein-DNA complexes. J. Mol. Biol. 2009, 390, 478–489. [Google Scholar] [CrossRef]
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G.; Murray, N.E. Highlights of the DNA cutters: A short history of the restriction enzymes. Nucleic Acids Res. 2014, 42, 3–19. [Google Scholar]
- Williams, R.J. Restriction endonucleases: Classification, properties, and applications. Mol. Biotechnol. 2003, 23, 225–243. [Google Scholar] [CrossRef]
- Pingoud, A.; Jeltsch, A. Structure and function of type II restriction endonucleases. Nucleic Acids Res. 2001, 29, 3705–3727. [Google Scholar] [CrossRef]
- Watanabe, N.; Takasaki, Y.; Sato, C.; Ando, S.; Tanaka, I. Structures of restriction endonuclease HindIII in complex with its cognate DNA and divalent cations. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 1326–1333. [Google Scholar] [CrossRef]
- Kostrewa, D.; Winkler, F.K. Mg2+ binding to the active site of EcoRV endonuclease: A crystallographic study of complexes with substrate and product DNA at 2 A resolution. Biochemistry 1995, 34, 683–696. [Google Scholar] [CrossRef]
- Roszczyk, E.; Goodgal, S. Methylase activities from Haemophilus influenzae that protect Haemophilus parainfluenzae transforming deoxyribonucleic acid from inactivation by Haemophilus influenzae endonuclease R. J. Bacteriol. 1975, 123, 287–293. [Google Scholar]
- Nwankwo, D.O.; Moran, L.S.; Slatko, B.E.; Waite-Rees, P.A.; Dorner, L.F.; Benner, J.S.; Wilson, G.G. Cloning, analysis and expression of the HindIII R-M-encoding genes. Gene 1994, 150, 75–80. [Google Scholar] [CrossRef]
- Martin, A.M.; Sam, M.D.; Reich, N.O.; Perona, J.J. Structural and energetic origins of indirect readout in site-specific DNA cleavage by a restriction endonuclease. Nat. Struct. Biol. 1999, 6, 269–277. [Google Scholar]
- Martin, A.M.; Horton, N.C.; Lusetti, S.; Reich, N.O.; Perona, J.J. Divalent metal dependence of site-specific DNA binding by EcoRV endonuclease. Biochemistry 1999, 38, 8430–8439. [Google Scholar]
- Otwinowski, Z.; Schevitz, R.W.; Zhang, R.G.; Lawson, C.L.; Joachimiak, A.; Marmorstein, R.Q.; Luisi, B.F.; Sigler, P.B. Crystal structure of trp repressor/operator complex at atomic resolution. Nature 1988, 335, 321–329. [Google Scholar] [CrossRef]
- Smith, S.A.; Rajur, S.B.; McLaughlin, L.W. Specific purine N7-nitrogens are critical for high affinity binding by the Trp repressor. Nat. Struct. Biol. 1994, 1, 198. [Google Scholar] [CrossRef]
- Zhang, A.P.; Pigli, Y.Z.; Rice, P.A. Structure of the LexA-DNA complex and implications for SOS box measurement. Nature 2010, 466, 883–886. [Google Scholar] [CrossRef]
- Fernandez De Henestrosa, A.R.; Ogi, T.; Aoyagi, S.; Chafin, D.; Hayes, J.J.; Ohmori, H.; Woodgate, R. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol. Microbiol. 2000, 35, 1560–1572. [Google Scholar]
- Wade, J.T.; Reppas, N.B.; Church, G.M.; Struhl, K. Genomic analysis of LexA binding reveals the permissive nature of the Escherichia coli genome and identifies unconventional target sites. Genes Dev. 2005, 19, 2619–2630. [Google Scholar] [CrossRef]
- White, R.J.; Jackson, S.P. The TATA-binding protein: A central role in transcription by RNA polymerases I, II and III. Trends Genet. 1992, 8, 284–288. [Google Scholar] [CrossRef]
- Sharp, P.A. TATA-binding protein is a classless factor. Cell 1992, 68, 819–821. [Google Scholar] [CrossRef]
- Rigby, P.W. Three in one and one in three: It all depends on TBP. Cell 1993, 72, 7–10. [Google Scholar] [CrossRef]
- Comai, L.; Tanese, N.; Tjian, R. The TATA-binding protein and associated factors are integral components of the RNA polymerase I transcription factor, SL1. Cell 1992, 68, 965–976. [Google Scholar] [CrossRef]
- Kim, J.L.; Burley, S.K. 1.9 A resolution refined structure of TBP recognizing the minor groove of TATAAAAG. Nat. Struct. Biol. 1994, 1, 638–653. [Google Scholar] [CrossRef]
- Kim, Y.; Geiger, J.H.; Hahn, S.; Sigler, P.B. Crystal structure of a yeast TBP/TATA-box complex. Nature 1993, 365, 512–520. [Google Scholar] [CrossRef]
- Arfin, S.M.; Long, A.D.; Ito, E.T.; Tolleri, L.; Riehle, M.M.; Paegle, E.S.; Hatfield, G.W. Global gene expression profiling in Escherichia coli K12. The effects of integration host factor. J. Biol. Chem. 2000, 275, 29672–29684. [Google Scholar]
- Kobryn, K.; Lavoie, B.D.; Chaconas, G. Supercoiling-dependent site-specific binding of HU to naked Mu DNA. J. Mol. Biol. 1999, 289, 777–784. [Google Scholar] [CrossRef]
- Hwang, D.S.; Kornberg, A. Opening of the replication origin of Escherichia coli by DnaA protein with protein HU or IHF. J. Biol. Chem. 1992, 267, 23083–23086. [Google Scholar]
- Kur, J.; Hasan, N.; Szybalski, W. Physical and biological consequences of interactions between integration host factor (IHF) and coliphage lambda late p'R promoter and its mutants. Gene 1989, 81, 1–15. [Google Scholar]
- Hillyard, D.R.; Edlund, M.; Hughes, K.T.; Marsh, M.; Higgins, N.P. Subunit-specific phenotypes of Salmonella typhimurium HU mutants. J. Bacteriol. 1990, 172, 5402–5407. [Google Scholar]
- Li, S.; Waters, R. Escherichia coli strains lacking protein HU are UV sensitive due to a role for HU in homologous recombination. J. Bacteriol. 1998, 180, 3750–3756. [Google Scholar]
- Miyabe, I.; Zhang, Q.M.; Kano, Y.; Yonei, S. Histone-like protein HU is required for recA gene-dependent DNA repair and SOS induction pathways in UV-irradiated Escherichia coli. Int. J. Radiat. Biol. 2000, 76, 43–49. [Google Scholar] [CrossRef]
- Johnson, R.C.; Johnson, L.M.; Schmidt, J.; Garder, J.F. The major nucleoid proteins in the structure and function of the Escherichia coli chromosome. In The Bacterial Chromosomes; ASM Press: Washington, DC, USA, 2005; Volume 1, pp. 65–132. [Google Scholar]
- Pil, P.M.; Chow, C.S.; Lippard, S.J. High-mobility-group 1 protein mediates DNA bending as determined by ring closures. Proc. Natl. Acad. Sci. USA 1993, 90, 9465–9469. [Google Scholar] [CrossRef]
- Paull, T.T.; Haykinson, M.J.; Johnson, R.C. The nonspecific DNA-binding and -bending proteins HMG1 and HMG2 promote the assembly of complex nucleoprotein structures. Genes Dev. 1993, 7, 1521–1534. [Google Scholar] [CrossRef]
- Shimizu, M.; Miyake, M.; Kanke, F.; Matsumoto, U.; Shindo, H. Characterization of the binding of HU and IHF, homologous histone-like proteins of Escherichia coli, to curved and uncurved DNA. Biochim. Biophys. Acta 1995, 1264, 330–336. [Google Scholar] [CrossRef]
- Swinger, K.K.; Rice, P.A. Structure-based analysis of HU-DNA binding. J. Mol. Biol. 2007, 365, 1005–1016. [Google Scholar] [CrossRef]
- Lynch, T.W.; Read, E.K.; Mattis, A.N.; Gardner, J.F.; Rice, P.A. Integration host factor: Putting a twist on protein-DNA recognition. J. Mol. Biol. 2003, 330, 493–502. [Google Scholar] [CrossRef]
- Dhavan, G.M.; Crothers, D.M.; Chance, M.R.; Brenowitz, M. Concerted binding and bending of DNA by Escherichia coli integration host factor. J. Mol. Biol. 2002, 315, 1027–1037. [Google Scholar]
- Bosch, D.; Campillo, M.; Pardo, L. Binding of proteins to the minor groove of DNA: What are the structural and energetic determinants for kinking a basepair step? J. Comput. Chem. 2003, 24, 682–691. [Google Scholar] [CrossRef]
- Grove, A.; Saavedra, T.C. The role of surface-exposed lysines in wrapping DNA about the bacterial histone-like protein HU. Biochemistry 2002, 41, 7597–7603. [Google Scholar] [CrossRef]
- Holbrook, J.A.; Tsodikov, O.V.; Saecker, R.M.; Record, M.T. Specific and non-specific interactions of integration host factor with DNA: Thermodynamic evidence for disruption of multiple IHF surface salt-bridges coupled to DNA binding. J. Mol. Biol. 2001, 310, 379–401. [Google Scholar] [CrossRef]
- Lowary, P.T.; Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998, 276, 19–42. [Google Scholar] [CrossRef]
- Satchwell, S.C.; Drew, H.R.; Travers, A.A. Sequence periodicities in chicken nucleosome core DNA. J. Mol. Biol. 1986, 191, 659–675. [Google Scholar] [CrossRef]
- Dayn, A.; Malkhosyan, S.; Mirkin, S.M. Transcriptionally driven cruciform formation in vivo. Nucleic Acids Res. 1992, 20, 5991–5997. [Google Scholar]
- Lobachev, K.S.; Rattray, A.; Narayanan, V. Hairpin- and cruciform-mediated chromosome breakage: Causes and consequences in eukaryotic cells. Front. Biosci. 2007, 12, 4208–4220. [Google Scholar]
- Maher, R.L.; Branagan, A.M.; Morrical, S.W. Coordination of DNA replication and recombination activities in the maintenance of genome stability. J. Cell. Biochem. 2011, 112, 2672–2682. [Google Scholar]
- Xu, J.; de Zhu, J.; Ni, M.; Wan, F.; Gu, J.R. The ATF/CREB site is the key element for transcription of the human RNA methyltransferase like 1(RNMTL1) gene, a newly discovered 17p13.3 gene. Cell Res. 2002, 12, 177–197. [Google Scholar] [CrossRef]
- Hanke, J.H.; Hambor, J.E.; Kavathas, P. Repetitive Alu elements form a cruciform structure that regulates the function of the human CD8 alpha T cell-specific enhancer. J. Mol. Biol. 1995, 246, 63–73. [Google Scholar] [CrossRef]
- Van Brabant, A.J.; Stan, R.; Ellis, N.A. DNA helicases, genomic instability, and human genetic disease. Annu. Rev. Genomics Hum. Genet. 2000, 1, 409–459. [Google Scholar] [CrossRef]
- Alvarez, D.; Novac, O.; Callejo, M.; Ruiz, M.T.; Price, G.B.; Zannis-Hadjopoulos, M. 14-3-3sigma is a cruciform DNA binding protein and associates in vivo with origins of DNA replication. J. Cell. Biochem. 2002, 87, 194–207. [Google Scholar]
- Callejo, M.; Alvarez, D.; Price, G.B.; Zannis-Hadjopoulos, M. The 14-3-3 protein homologues from Saccharomyces cerevisiae, Bmh1p and Bmh2p, have cruciform DNA-binding activity and associate in vivo with ARS307. J. Biol. Chem. 2002, 277, 38416–38423. [Google Scholar]
- Zannis-Hadjopoulos, M.; Yahyaoui, W.; Callejo, M. 14-3-3 cruciform-binding proteins as regulators of eukaryotic DNA replication. Trends Biochem. Sci. 2008, 33, 44–50. [Google Scholar] [CrossRef]
- Wakasugi, M.; Reardon, J.T.; Sancar, A. The non-catalytic function of XPG protein during dual incision in human nucleotide excision repair. J. Biol. Chem. 1997, 272, 16030–16034. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, C.J.; Arunkumar, A.I.; Chazin, W.J.; Choi, B.S. NMR study on the interaction between RPA and DNA decamer containing cis-syn cyclobutane pyrimidine dimer in the presence of XPA: Implication for damage verification and strand-specific dual incision in nucleotide excision repair. Nucleic Acids Res. 2003, 31, 4747–4754. [Google Scholar] [CrossRef]
- Sekelsky, J.J.; Hollis, K.J.; Eimerl, A.I.; Burtis, K.C.; Hawley, R.S. Nucleotide excision repair endonuclease genes in Drosophila melanogaster. Mutat. Res. 2000, 459, 219–228. [Google Scholar]
- Stefanovsky, V.Y.; Moss, T. The cruciform DNA mobility shift assay: A tool to study proteins that recognize bent DNA. Methods Mol. Biol. 2009, 543, 537–546. [Google Scholar] [CrossRef]
- Van Houte, L.P.; Chuprina, V.P.; van der Wetering, M.; Boelens, R.; Kaptein, R.; Clevers, H. Solution structure of the sequence-specific HMG box of the lymphocyte transcriptional activator Sox-4. J. Biol. Chem. 1995, 270, 30516–30524. [Google Scholar]
- Pearson, C.E.; Ruiz, M.T.; Price, G.B.; Zannis-Hadjopoulos, M. Cruciform DNA binding protein in HeLa cell extracts. Biochemistry 1994, 33, 14185–14196. [Google Scholar] [CrossRef]
- Declais, A.C.; Lilley, D.M. New insight into the recognition of branched DNA structure by junction-resolving enzymes. Curr. Opin. Struct. Biol. 2008, 18, 86–95. [Google Scholar] [CrossRef]
- Lilley, D.M.; White, M.F. The junction-resolving enzymes. Nat. Rev. Mol. Cell. Biol. 2001, 2, 433–443. [Google Scholar]
- Khuu, P.A.; Voth, A.R.; Hays, F.A.; Ho, P.S. The stacked-X DNA Holliday junction and protein recognition. J. Mol. Recognit. 2006, 19, 234–242. [Google Scholar] [CrossRef]
- Lilley, D.M. Structures of helical junctions in nucleic acids. Q. Rev. Biophys. 2000, 33, 109–159. [Google Scholar] [CrossRef]
- Brill, S.J. Linking the enzymes that unlink DNA. Mol. Cell. 2013, 52, 159–160. [Google Scholar] [CrossRef]
- Brazda, V.; Laister, R.C.; Jagelska, E.B.; Arrowsmith, C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011, 12, 1–16. [Google Scholar] [CrossRef]
- Gorecka, K.M.; Komorowska, W.; Nowotny, M. Crystal structure of RuvC resolvase in complex with Holliday junction substrate. Nucleic Acids Res. 2013, 41, 9945–9955. [Google Scholar] [CrossRef]
- Biertumpfel, C.; Yang, W.; Suck, D. Crystal structure of T4 endonuclease VII resolving a Holliday junction. Nature 2007, 449, 616–620. [Google Scholar] [CrossRef]
- Ortiz-Lombardia, M.; Gonzalez, A.; Eritja, R.; Aymami, J.; Azorin, F.; Coll, M. Crystal structure of a DNA Holliday junction. Nat. Struct. Biol. 1999, 6, 913–917. [Google Scholar] [CrossRef]
- Mashal, R.D.; Koontz, J.; Sklar, J. Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases. Nat. Genet. 1995, 9, 177–183. [Google Scholar] [CrossRef]
- Hadden, J.M.; Declais, A.C.; Carr, S.B.; Lilley, D.M.; Phillips, S.E. The structural basis of Holliday junction resolution by T7 endonuclease I. Nature 2007, 449, 621–624. [Google Scholar] [CrossRef]
- Declais, A.C.; Liu, J.; Freeman, A.D.; Lilley, D.M. Structural recognition between a four-way DNA junction and a resolving enzyme. J. Mol. Biol. 2006, 359, 1261–1276. [Google Scholar] [CrossRef]
- Freeman, A.D.; Declais, A.C.; Lilley, D.M. The importance of the N-terminus of T7 endonuclease I in the interaction with DNA junctions. J. Mol. Biol. 2013, 425, 395–410. [Google Scholar] [CrossRef]
- Herbert, A.; Alfken, J.; Kim, Y.G.; Mian, I.S.; Nishikura, K.; Rich, A.A. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc. Natl. Acad. Sci. USA 1997, 94, 8421–8426. [Google Scholar]
- Herbert, A.G.; Spitzner, J.R.; Lowenhaupt, K.; Rich, A. Z-DNA binding protein from chicken blood nuclei. Proc. Natl. Acad. Sci. USA 1993, 90, 3339–3342. [Google Scholar] [CrossRef]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef]
- Schwartz, T.; Rould, M.A.; Lowenhaupt, K.; Herbert, A.; Rich, A. Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science 1999, 284, 1841–1845. [Google Scholar] [CrossRef]
- De Rosa, M.; de Sanctis, D.; Rosario, A.L.; Archer, M.; Rich, A.; Athanasiadis, A.; Carrondo, M.A. Crystal structure of a junction between two Z-DNA helices. Proc. Natl. Acad. Sci. USA 2010, 107, 9088–9092. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Harteis, S.; Schneider, S. Making the Bend: DNA Tertiary Structure and Protein-DNA Interactions. Int. J. Mol. Sci. 2014, 15, 12335-12363. https://doi.org/10.3390/ijms150712335
Harteis S, Schneider S. Making the Bend: DNA Tertiary Structure and Protein-DNA Interactions. International Journal of Molecular Sciences. 2014; 15(7):12335-12363. https://doi.org/10.3390/ijms150712335
Chicago/Turabian StyleHarteis, Sabrina, and Sabine Schneider. 2014. "Making the Bend: DNA Tertiary Structure and Protein-DNA Interactions" International Journal of Molecular Sciences 15, no. 7: 12335-12363. https://doi.org/10.3390/ijms150712335