Induction of Apurinic Endonuclease 1 Overexpression by Endoplasmic Reticulum Stress in Hepatoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
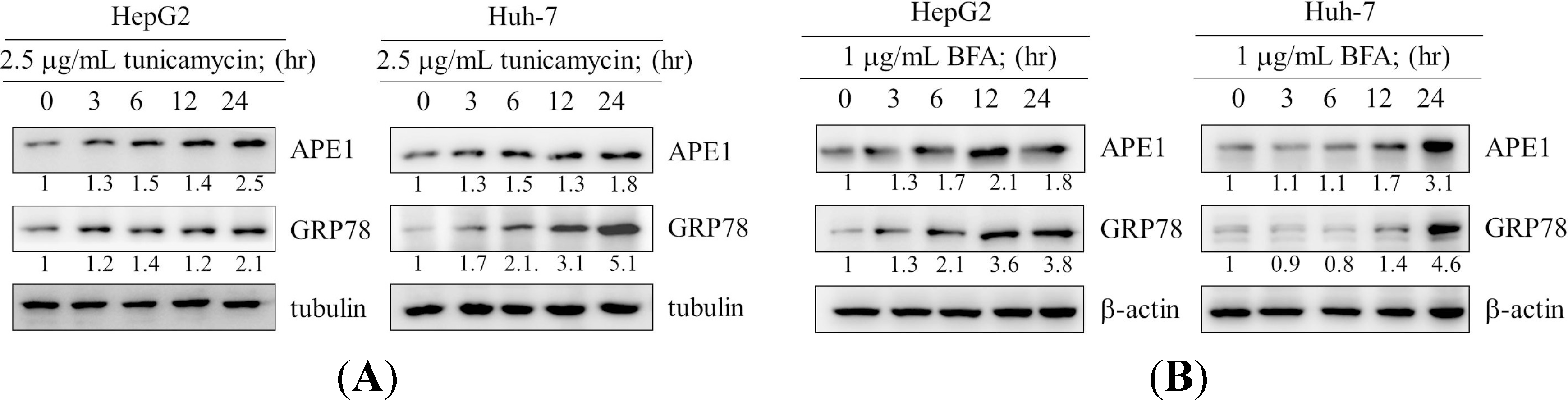
2.1. Induction of Apurinic Endonuclease 1 (APE1) Expression by Endoplasmic Reticulum (ER) Stress
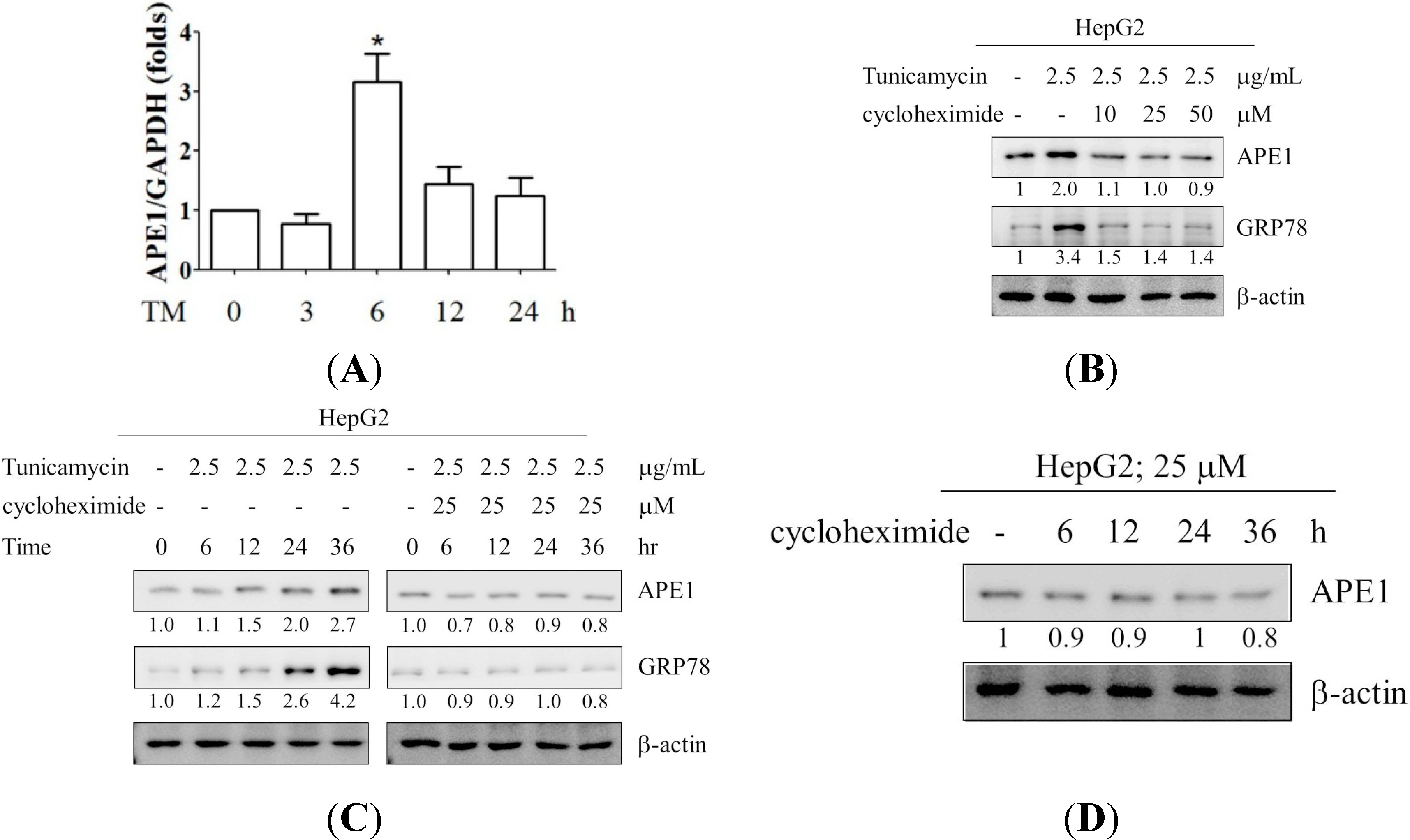
2.2. Induction of APE1 Expression Is Transcription-Dependent


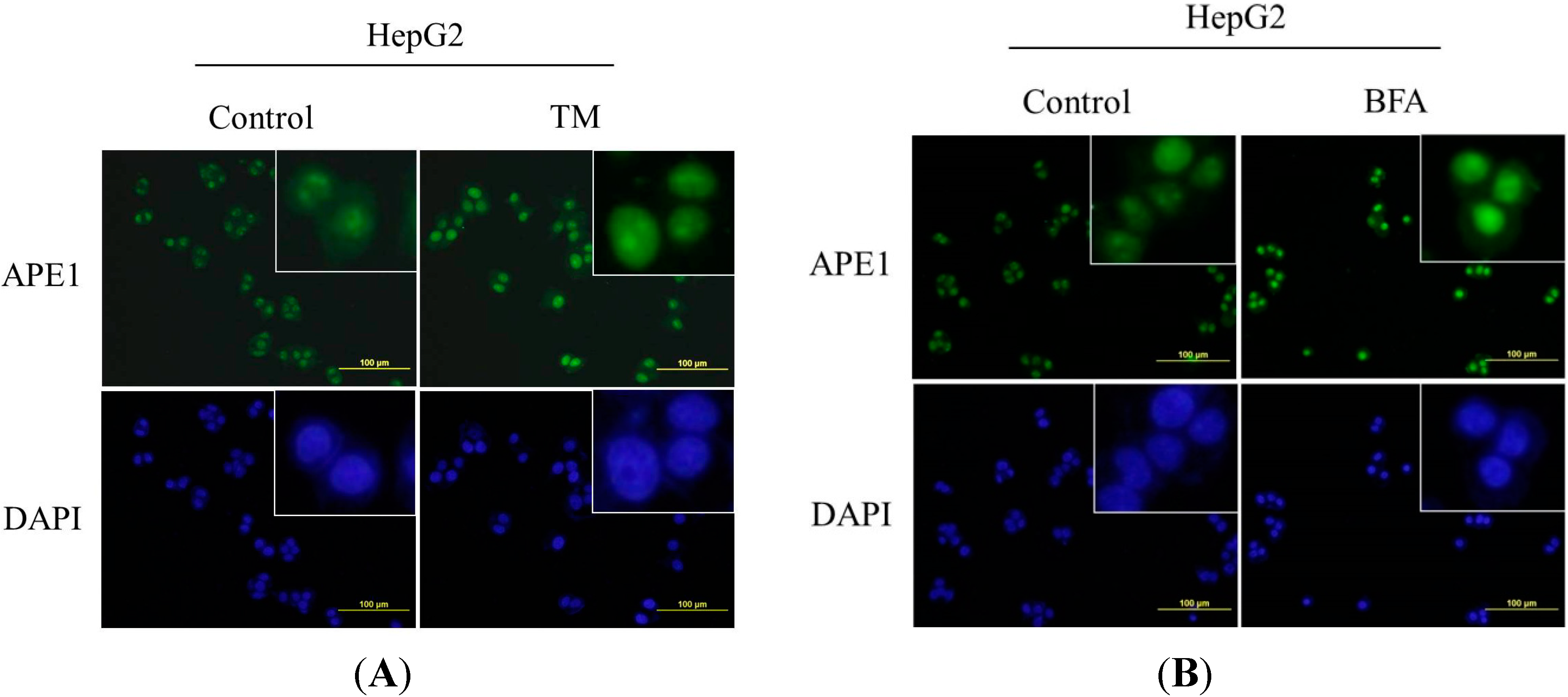
2.3. Nuclear Localization of APE1 during ER Stress

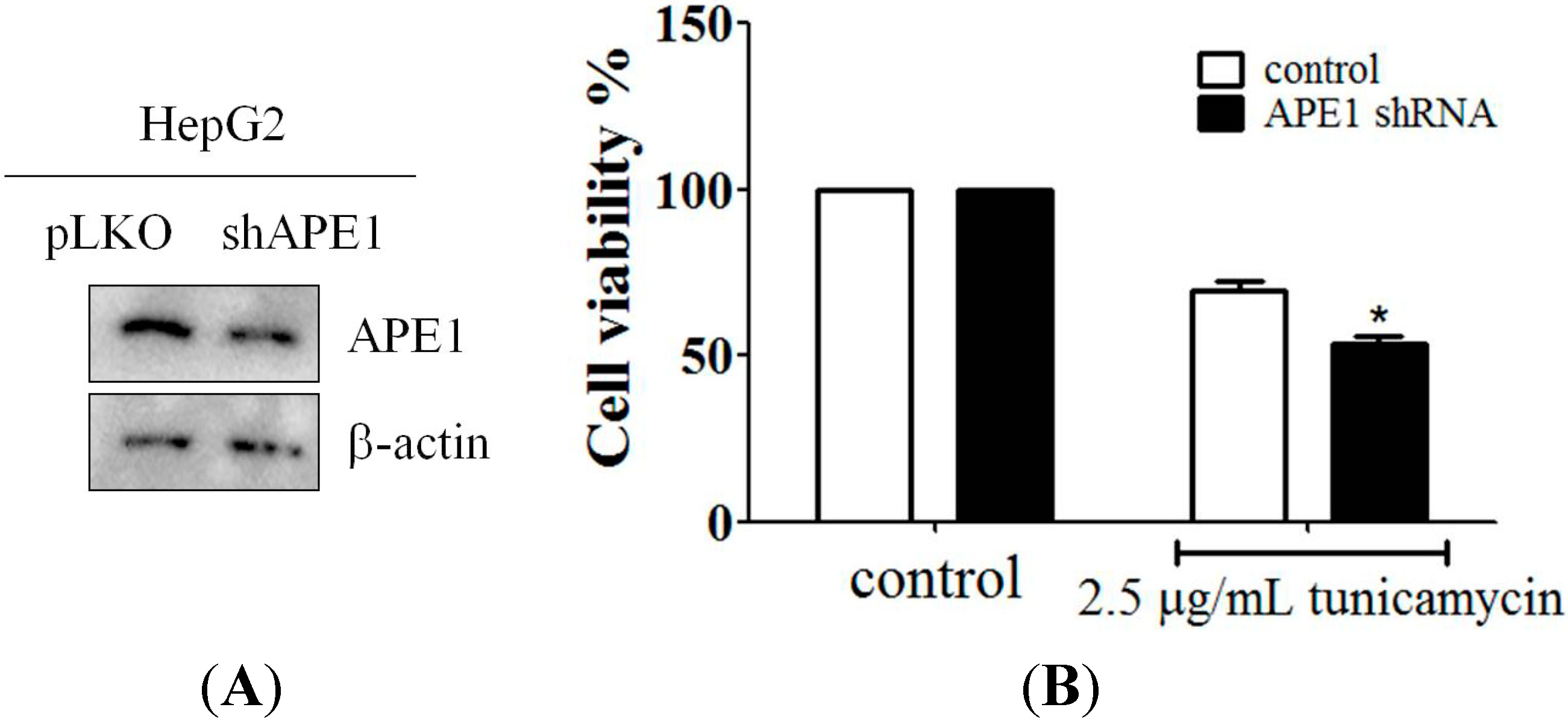
2.4. Downregulation of APE1 Expression Reduced Cell Survival in Response to ER Stress
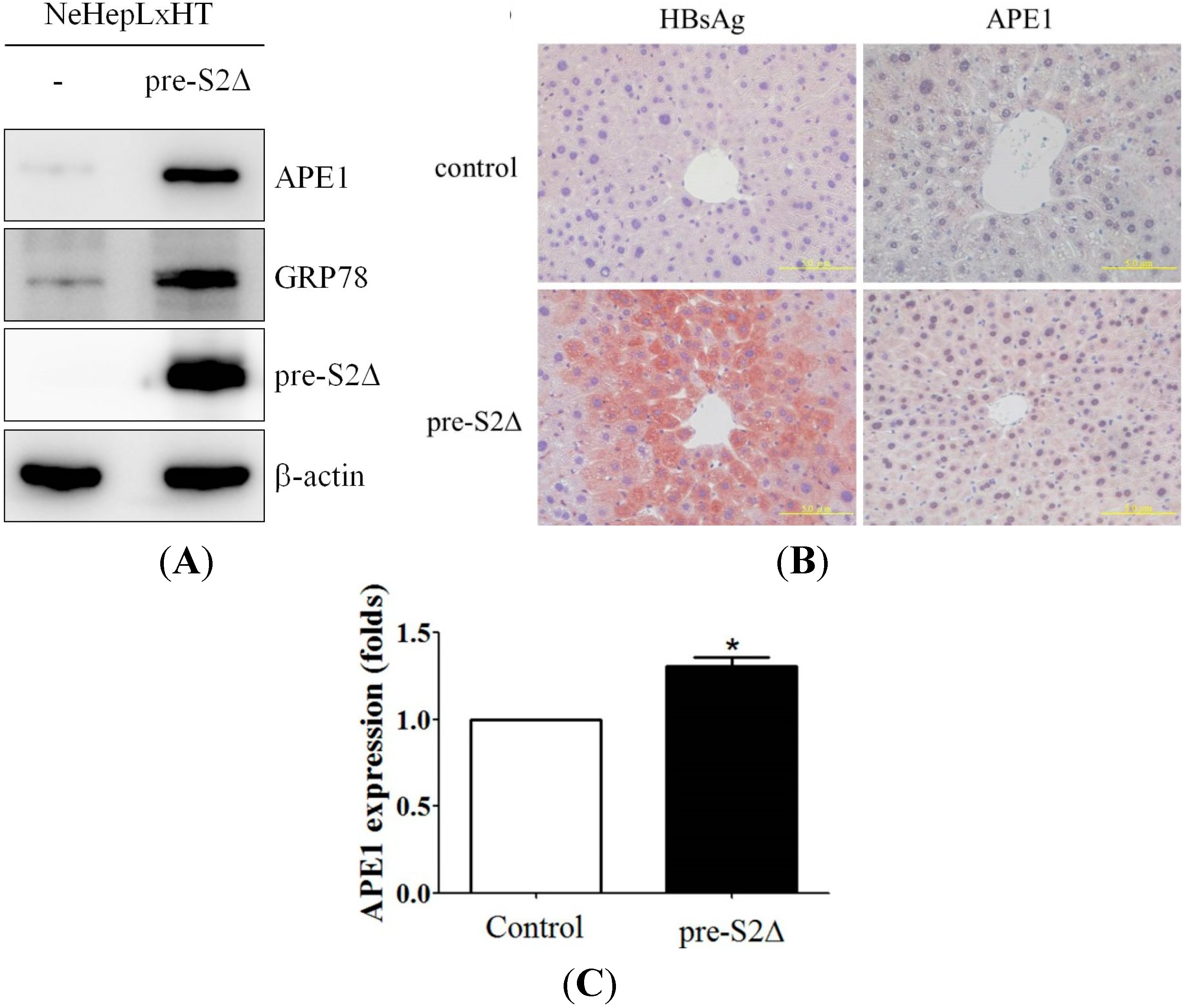
2.5. Hepatitis B Virus Mutant Large Surface Protein Can Induce APE1 Expression in Vitro and in Vivo

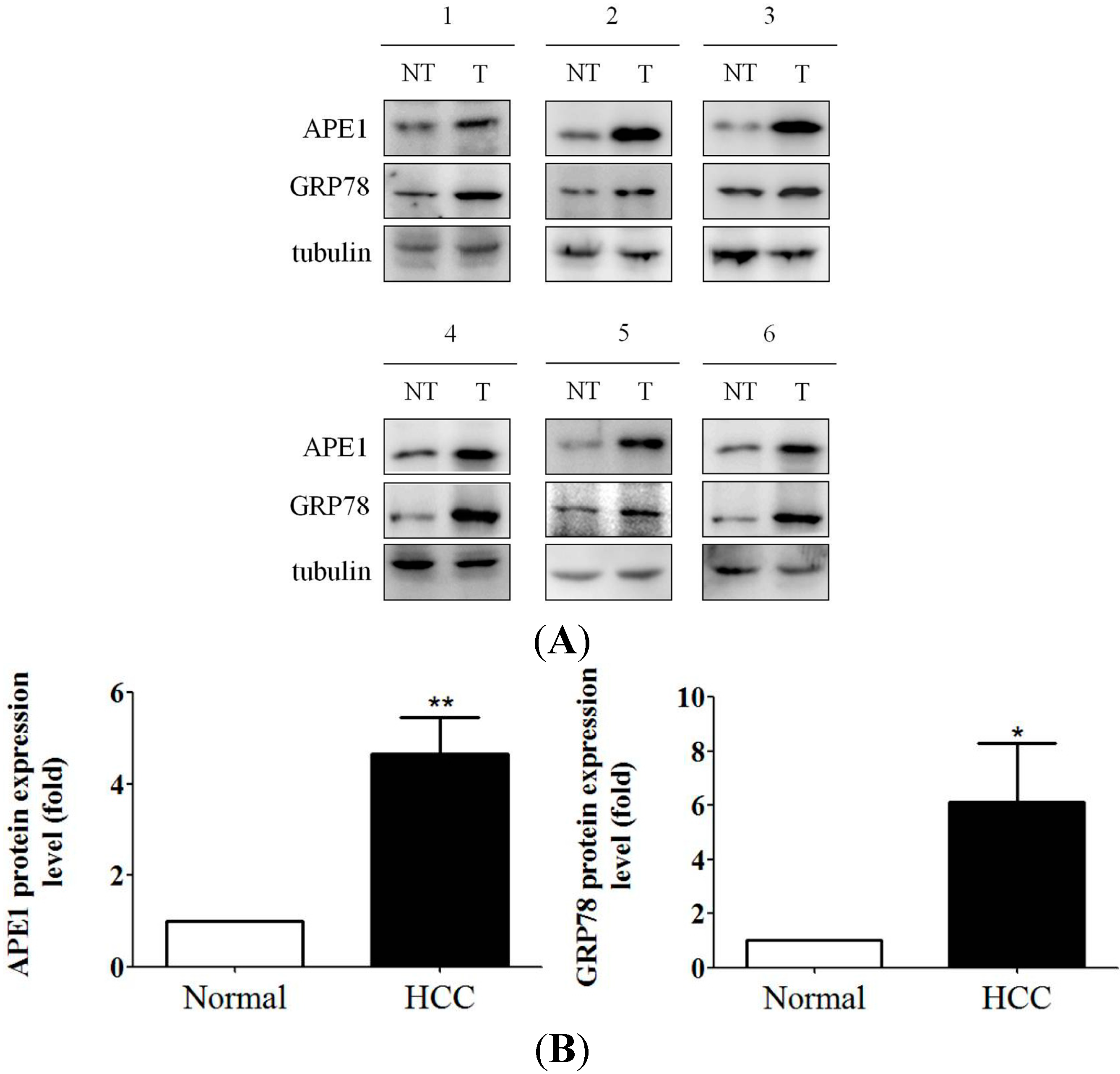
2.6. Increased APE1 Expression Is Observed in ER Stress-Associated Liver Tumor Tissues

3. Discussion

4. Experimental Section
4.1. Cell Culture and Material
4.2. Western Blot Analysis
4.3. MTT Assay
4.4. Real-Time PCR
4.5. Transfection of pLKO-APE1 shRNA Vector
4.6. Histological Analysis of Pre-S2Δ Transgenic Mice Liver Tissue
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Kao, J.H.; Chen, D.S. Changing disease burden of hepatocellular carcinoma in the Far East and Southeast Asia. Liv. Int. 2005, 25, 696–703. [Google Scholar] [CrossRef]
- Jeannot, E.; Boorman, G.A.; Kosyk, O.; Bradford, B.U.; Shymoniak, S.; Tumurbaatar, B.; Weinman, S.A.; Melnyk, S.B.; Tryndyak, V.; Pogribny, I.P.; et al. Increased incidence of aflatoxin B1-induced liver tumors in hepatitis virus C transgenic mice. Int. J. Cancer 2012, 130, 1347–1356. [Google Scholar]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef]
- Lee, J.W.; Soung, Y.H.; Kim, S.Y.; Lee, H.W.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene 2005, 24, 1477–1480. [Google Scholar]
- Feitelson, M.A.; Sun, B.; Satiroglu Tufan, N.L.; Liu, J.; Pan, J.; Lian, Z. Genetic mechanisms of hepatocarcinogenesis. Oncogene 2002, 21, 2593–2604. [Google Scholar]
- Evans, A.A.; London, W.T.; Gish, R.G.; Cohen, C.; Block, T.M. Chronic HBV infection outside treatment guidelines: Is treatment needed? Antivir. Ther. 2013, 18, 229–235. [Google Scholar]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef]
- Wong, V.W.; Chan, H.L. Prevention of hepatocellular carcinoma: A concise review of contemporary issues. Ann. Hepatol. 2012, 11, 284–293. [Google Scholar]
- Fan, Y.F.; Lu, C.C.; Chang, Y.C.; Chang, T.T.; Lin, P.W.; Lei, H.Y.; Su, I.J. Identification of a pre-S2 mutant in hepatocytes expressing a novel marginal pattern of surface antigen in advanced diseases of chronic hepatitis B virus infection. J. Gastroenterol. Hepatol. 2000, 15, 519–528. [Google Scholar]
- Fan, Y.F.; Lu, C.C.; Chen, W.C.; Yao, W.J.; Wang, H.C.; Chang, T.T.; Lei, H.Y.; Shiau, A.L.; Su, I.J. Prevalence and significance of hepatitis B virus (HBV) pre-S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 2001, 33, 277–286. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Su, I.J.; Yen, C.J.; Tsai, T.F.; Tsai, H.W.; Tsai, H.N.; Huang, Y.J.; Chen, Y.Y.; Ai, Y.L.; Kao, L.Y.; et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid suppresses the pro-oncogenic effects induced by hepatitis B virus pre-S2 mutant oncoprotein and represents a potential chemopreventive agent in high-risk chronic HBV patients. Carcinogenesis 2013, 34, 475–485. [Google Scholar]
- Lin, C.M.; Wang, G.M.; Jow, G.M.; Chen, B.F. Functional analysis of hepatitis B virus pre-S deletion variants associated with hepatocellular carcinoma. J. Biomed. Sci. 2012, 19, 17. [Google Scholar] [CrossRef]
- Chou, K.M.; Cheng, Y.C. An exonucleolytic activity of human apurinic/apyrimidinic endonuclease on 3'mispaired DNA. Nature 2002, 415, 655–659. [Google Scholar] [CrossRef]
- Ishchenko, A.A.; Yang, X.; Ramotar, D.; Saparbaev, M. The 3'->5' exonuclease of Apn1 provides an alternative pathway to repair 7,8-dihydro-8-oxodeoxyguanosine in Saccharomyces cerevisiae. Mol. Cell. Biol. 2005, 25, 6380–6390. [Google Scholar] [CrossRef]
- Li, Y.; Liu, X.; Zhou, T.; Kelley, M.R.; Edwards, P.; Gao, H.; Qiao, X. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014, 2, 485–494. [Google Scholar]
- Evans, A.R.; Limp-Foster, M.; Kelley, M.R. Going APE over ref-1. Mutat. Res. 2000, 461, 83–108. [Google Scholar]
- Tell, G.; Damante, G.; Caldwell, D.; Kelley, M.R. The intracellular localization of APE1/Ref-1: More than a passive phenomenon? Antioxid. Redox Signal. 2005, 7, 367–384. [Google Scholar] [CrossRef]
- Pines, A.; Perrone, L.; Bivi, N.; Romanello, M.; Damante, G.; Gulisano, M.; Kelley, M.R.; Quadrifoglio, F.; Tell, G. Activation of APE1/ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res. 2005, 33, 4379–4394. [Google Scholar] [CrossRef]
- Robertson, K.A.; Bullock, H.A.; Xu, Y.; Tritt, R.; Zimmerman, E.; Ulbright, T.M.; Foster, R.S.; Einhorn, L.H.; Kelley, M.R. Altered expression of APE1/ref-1 in germ cell tumors and over-expression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001, 61, 2220–2225. [Google Scholar]
- Thomson, B.; Tritt, R.; Davis, M.; Kelley, M.R. Histology-specific expression of a DNA repair protein in pediatric rhabdomyosarcomas. J. Pediatr. Hematol. Oncol. 2001, 23, 234–239. [Google Scholar] [CrossRef]
- Puglisi, F.; Barbone, F.; Tell, G.; Aprile, G.; Pertoldi, B.; Raiti, C.; Kelley, M.R.; Damante, G.; Sobrero, A.; Beltrami, C.A.; et al. Prognostic role of APE1/ref-1 subcellular expression in stage I–III breast carcinomas. Oncol. Rep. 2002, 9, 11–7. [Google Scholar]
- Frau, M.; Feo, F.; Pascale, R.M. Pleiotropic effects of methionine adenosyltransferases deregulation as determinants of liver cancer progression and prognosis. J. Hepatol. 2013, 59, 830–841. [Google Scholar]
- Wang, D.; Xiang, D.B.; Yang, X.Q.; Chen, L.S.; Li, M.X.; Zhong, Z.Y.; Zhang, Y.S. APE1 over-expression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer 2009, 66, 298–304. [Google Scholar]
- Londero, A.P.; Orsaria, M.; Tell, G.; Marzinotto, S.; Capodicasa, V.; Poletto, M.; Vascotto, C.; Sacco, C.; Mariuzzi, L. Expression and prognostic significance of APE1/Ref-1 and NPM1 proteins in high-grade ovarian serous cancer. Am. J. Clin. Pathol. 2014, 141, 404–414. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, S.; Sandusky, G.E.; Kelley, M.R.; Fishel, M.L. Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Investig. 2010, 28, 885–895. [Google Scholar] [CrossRef]
- Kothandapani, A.; Dangeti, V.S.; Brown, A.R.; Banze, L.A.; Wang, X.H.; Sobol, R.W.; Patrick, S.M. Novel role of base excision repair in mediating cisplatin cytotoxicity. J. Biol. Chem. 2011, 286, 14564–14574. [Google Scholar]
- Sanz-Pamplona, R.; Aragüés, R.; Driouch, K.; Martín, B.; Oliva, B.; Gil, M.; Boluda, S.; Fernández, P.L.; Martínez, A.; Moreno, V. Expression of endoplasmic reticulum stress proteins is a candidate marker of brain metastasis in both ErbB-2+ and ErbB-2− primary breast tumors. Am. J. Pathol. 2011, 179, 564–579. [Google Scholar] [CrossRef]
- Kim, K.M.; Yu, T.K.; Chu, H.H.; Park, H.S.; Jang, K.Y.; Moon, W.S.; Kang, M.J. Lee, D.G.; Kim, M.H.; Lee, J.H.; et al. Expression of ER stress and autophagy-related molecules in human non-small cell lung cancer and premalignant lesions. Int. J. Cancer 2012, 131, E362–E370. [Google Scholar]
- Fang, D.L.; Wan, Y.; Shen, W.; Cao, J.; Sun, Z.X.; Yu, H.H.; Zhang, Q.; Cheng, W.H.; Chen, J.; Ning, B. Endoplasmic reticulum stress leads to lipid accumulation through up-regulation of SREBP-1c in normal hepatic and hepatoma cells. Mol. Cell. Biochem. 2013, 381, 127–137. [Google Scholar] [CrossRef]
- De la Cadena, S.G.; Hernández-Fonseca, K.; Camacho-Arroyo, I.; Massieu, L. Glucose deprivation induces reticulum stress by the PERK pathway and caspase-7- and calpain-mediated caspase-12 activation. Apoptosis 2014, 19, 414–427. [Google Scholar] [CrossRef]
- Chen, P.; Burdette, A.J.; Porter, J.C.; Ricketts, J.C.; Fox, S.A.; Nery, F.C.; Hewett, J.W.; Berkowitz, L.A.; Breakefield, X.O.; Caldwell, K.A.; et al. The early-onset torsion dystonia-associated protein, torsinA, is a homeostatic regulator of endoplasmic reticulum stress response. Hum. Mol. Genet. 2010, 19, 3502–3515. [Google Scholar]
- Gass, J.N.; Jiang, H.Y.; Wek, R.C.; Brewer, J.W. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol. Immunol. 2008, 45, 1035–1043. [Google Scholar]
- Gass, J.N.; Gifford, N.M.; Brewer, J.W. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J. Biol. Chem. 2002, 277, 49047–49054. [Google Scholar]
- Hung, J.H.; Su, I.J.; Lei, H.Y.; Wang, H.C.; Lin, W.C.; Chang, W.T.; Huang, W.; Chang, W.C.; Chang, Y.S.; Chen, C.C.; et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-κB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 46384–46392. [Google Scholar]
- Merquiol, E.; Uzi, D.; Mueller, T.; Goldenberg, D.; Nahmias, Y.; Xavier, R.J.; Tirosh, B.; Shibolet, O. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One 2011, 6, e24660. [Google Scholar]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef]
- Xu, Z.; Yen, T.S. Intracellular retention of surface protein by a hepatitis B virus mutant that releases virion particles. J. Virol. 1996, 70, 133–140. [Google Scholar]
- Chen, B.F.; Liu, C.J.; Jow, G.M.; Chen, P.J.; Kao, J.H.; Chen, D.S. High prevalence and mapping of pre-S deletion in hepatitis B virus carriers with progressive liver diseases. Gastroenterology 2006, 130, 1153–1168. [Google Scholar] [CrossRef]
- Di Maso, V.; Avellini, C.; Crocè, L.S.; Rosso, N.; Quadrifoglio, F.; Cesaratto, L.; Codarin, E.; Bedogni, G.; Beltrami, C.A.; Tell, G.; et al. Subcellular localization of APE1/Ref-1 in human hepatocellular carcinoma: Possible prognostic significance. Mol. Med. 2007, 13, 89–96. [Google Scholar]
- Sak, S.C.; Harnden, P.; Johnston, C.F.; Paul, A.B.; Kiltie, A.E. APE1 and XRCC1 protein expression levels predict cancer-specific survival following radical radiotherapy in bladder cancer. Clin. Cancer Res. 2005, 11, 6205–6211. [Google Scholar]
- Puglisi, F.; Aprile, G.; Minisini, A.M.; Barbone, F.; Cataldi, P.; Tell, G.; Kelley, M.R.; Damante, G.; Beltrami, C.A.; di Loreto, C. Prognostic significance of APE1/ref-1 subcellular localization in non-small cell lung carcinomas. Anticancer. Res. 2001, 21, 4041–4049. [Google Scholar]
- Naidu, M.D.; Mason, J.M.; Pica, R.V.; Fung, H.; Peña, L.A. Radiation resistance in glioma cells determined by DNA damage repair activity of APE1/ref-1. J. Radiat. Res. 2010, 51, 393–404. [Google Scholar] [CrossRef]
- Wang, D.; Luo, M.; Kelley, M.R. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol. Cancer Ther. 2004, 3, 679–686. [Google Scholar]
- Cun, Y.; Zhang, Q.; Xiong, C.; Li, M.; Dai, N.; Zhang, S.; Wang, D. Combined use of adenoviral vector Ad5/F35-mediated APE1 siRNA enhances the therapeutic efficacy ofadenoviral-mediated p53 gene transfer in hepatoma cells in vitro and in vivo. Oncol. Rep. 2013, 29, 2197–2204. [Google Scholar]
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Chang, W.W.; Lei, H.Y.; Lai, M.D.; Chang, W.T.; Huang, W. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004, 25, 2023–2032. [Google Scholar] [CrossRef]
- Wang, H.C.; Chang, W.T.; Chang, W.W.; Wu, H.C.; Huang, W.; Lei, H.Y.; Lai, M.D.; Fausto, N.; Su, I.J. Hepatitis B virus pre-S2 mutant upregulates cyclin a expression and induces nodular proliferation of hepatocytes. Hepatology 2005, 41, 761–770. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Tsai, J.H.; Huang, Y.J.; Chang, W.W.; Lai, M.D.; Lei, H.Y.; Huang, W. Hepatitis B virus pre-S2 mutant surface antigen induces degradation of cyclin-dependent kinase inhibitor p27Kip1 through c-Jun activation domain-binding protein 1. Mol. Cancer Res. 2007, 5, 1063–1072. [Google Scholar] [CrossRef]
- Yang, J.C.; Teng, C.F.; Wu, H.C.; Tsai, H.W.; Chuang, H.C.; Tsai, T.F.; Hsu, Y.H.; Huang, W.; Wu, L.W.; Su, I.J. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology 2009, 49, 1962–1971. [Google Scholar] [CrossRef]
- Hung, J.H.; Yan, C.W.; Su, I.J.; Wang, H.C.; Lei, H.Y.; Lin, W.C.; Chang, W.T.; Huang, W.; Lu, T.J.; Lai, M.D. Hepatitis B virus surface antigen interacts with acid α-glucosidase and alters glycogen metabolism. Hepatol. Res. 2010, 40, 633–640. [Google Scholar] [CrossRef]
- Chang, Y.S.; Tsai, C.T.; Huangfu, C.A.; Huang, W.Y.; Lei, H.Y.; Lin, C.F.; Su, I.J.; Chang, W.T.; Wu, P.H.; Chen, Y.T.; et al. ACSL3 and GSK-3β are essential for lipid upregulation induced by endoplasmic reticulum stress in liver cells. J. Cell. Biochem. 2011, 112, 881–893. [Google Scholar] [CrossRef]
- Hung, J.H.; Teng, Y.N.; Wang, L.H.; Su, I.J.; Wang, C.C.; Huang, W.; Lee, K.H.; Lu, K.Y.; Wang, L.H. Induction of Bcl-2 expression by hepatitis B virus pre-S2 mutant large surface protein resistance to 5-fluorouracil treatment in Huh-7 cells. PLoS One 2011, 6, e28977. [Google Scholar]
- Yeo, W.; Mo, F.K.; Chan, S.L.; Leung, N.W.; Hui, P.; Lam, W.Y.; Mok, T.S.; Lam, K.C.; Ho, W.M.; Koh, J.; et al. Hepatitis B viral load predicts survival of HCC patients undergoing systemic chemotherapy. Hepatology 2007, 45, 1382–1389. [Google Scholar]
- Yeo, W.; Chan, P.K.; Ho, W.M.; Zee, B.; Lam, K.C.; Lei, K.I.; Chan, A.T.; Mok, T.S.; Lee, J.J.; Leung, T.W.; et al. Lamivudine for the prevention of hepatitis B virus reactivation in hepatitis B s-antigen seropositive cancer patients undergoing cytotoxic chemotherapy. J Clin. Oncol. 2004, 22, 927–934. [Google Scholar]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar]
- Xing, X.; Lai, M.; Wang, Y. Over-expression of glucose-regulated protein 78 in colon cancer. Clin. Chim. Acta 2006, 364, 308–315. [Google Scholar] [CrossRef]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar]
- Liu, C.H.; Chang, S.H.; Narko, K.; Trifan, O.C.; Wu, M.T.; Smith, E.; Haudenschild, C.; Lane, T.F.; Hla, T. Over-expression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J. Biol. Chem. 2001, 276, 18563–18569. [Google Scholar]
- Rahman, M.; Selvarajan, K.; Hasan, M.R.; Chan, A.P.; Jin, C.; Kim, J.; Chan, S.K.; Le, N.D.; Kim, Y.B.; Tai, I.T. Inhibition of COX-2 in colon cancer modulates tumor growth and MDR-1 expression to enhance tumor regression in therapy-refractory cancers in vivo. Neoplasia 2012, 14, 624–633. [Google Scholar]
- Sitia, G.; Aiolfi, R.; di Lucia, P.; Mainetti, M.; Fiocchi, A.; Mingozzi, F.; Esposito, A.; Ruggeri, Z.M.; Chisari, F.V.; Iannacone, M.; et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. USA 2012, 109, E2165–E2172. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cheng, T.-L.; Chen, P.-S.; Li, R.-H.; Yuan, S.-S.; Su, I.-J.; Hung, J.-H. Induction of Apurinic Endonuclease 1 Overexpression by Endoplasmic Reticulum Stress in Hepatoma Cells. Int. J. Mol. Sci. 2014, 15, 12442-12457. https://doi.org/10.3390/ijms150712442
Cheng T-L, Chen P-S, Li R-H, Yuan S-S, Su I-J, Hung J-H. Induction of Apurinic Endonuclease 1 Overexpression by Endoplasmic Reticulum Stress in Hepatoma Cells. International Journal of Molecular Sciences. 2014; 15(7):12442-12457. https://doi.org/10.3390/ijms150712442
Chicago/Turabian StyleCheng, Tsung-Lin, Pin-Shern Chen, Ren-Hao Li, Shyng-Shiou Yuan, Ih-Jen Su, and Jui-Hsiang Hung. 2014. "Induction of Apurinic Endonuclease 1 Overexpression by Endoplasmic Reticulum Stress in Hepatoma Cells" International Journal of Molecular Sciences 15, no. 7: 12442-12457. https://doi.org/10.3390/ijms150712442