Identifying the Subfamilies of Voltage-Gated Potassium Channels Using Feature Selection Technique


Abstract
:1. Introduction

2. Results and Discussion
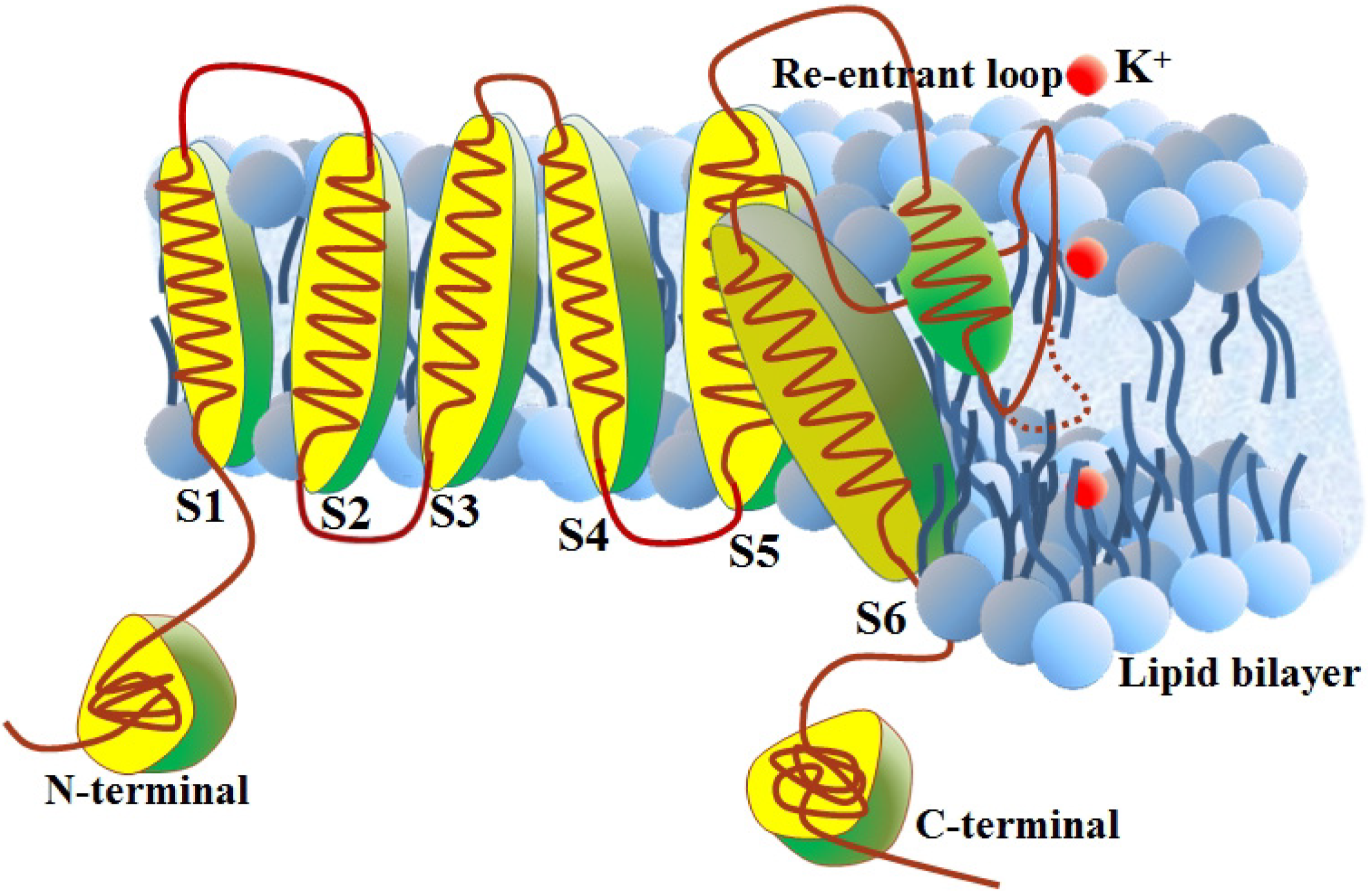
2.1. Benchmark Dataset

{kind=link}
{kind=link}
{kind=link}
| Dataset | Channel Subfamilies | Number of VKC Samples |
|---|---|---|
| S1 | Kv1 | 82 |
| S2 | Kv2 | 16 |
| S3 | Kv3 | 37 |
| S4 | Kv4 | 32 |
| S5 | Kv6 | 10 |
| S6 | Kv7 | 40 |
| S | Overall | 217 |
2.2. The Tripeptide Composition
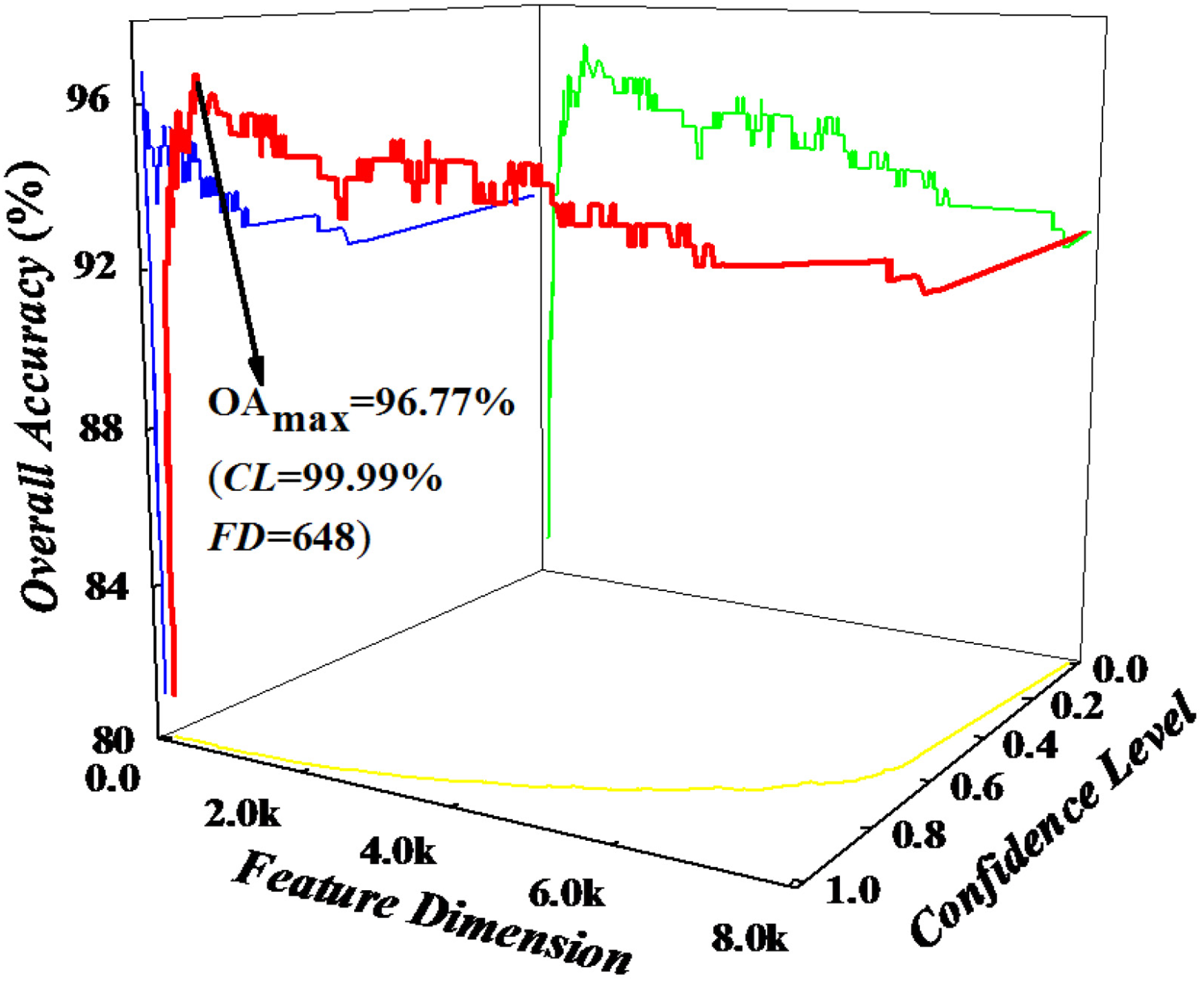
2.3. Feature Selection

 denotes the total occurrence number of all tripeptides in the benchmark dataset.
denotes the total occurrence number of all tripeptides in the benchmark dataset.  is the occurrence number of all tripeptides in the k-th VKC subfamily. The nik represents the number of the i-th tripeptides occurring in the k-th VKC subfamily. Correspondingly, the probability of the non-occurrence in the k-th VKC subfamily is defined as qk = 1 − pk.
is the occurrence number of all tripeptides in the k-th VKC subfamily. The nik represents the number of the i-th tripeptides occurring in the k-th VKC subfamily. Correspondingly, the probability of the non-occurrence in the k-th VKC subfamily is defined as qk = 1 − pk. represents the total occurrence number of the i-th tripeptide in benchmark dataset. That is to say, under the condition of the prior probability pk, one performs trial or observation with Ni times. We may calculate the posterior probability Pik of the i-th tripeptide occurring nik or more times in the k-th VKC subfamily as following:
represents the total occurrence number of the i-th tripeptide in benchmark dataset. That is to say, under the condition of the prior probability pk, one performs trial or observation with Ni times. We may calculate the posterior probability Pik of the i-th tripeptide occurring nik or more times in the k-th VKC subfamily as following:

2.4. Support Vector Machine

 is the i-th training vector. The yi represents the type of the i-th training vector. αi is coefficient which can be solved by quadratic programming. The b is the intercept parameter.
is the i-th training vector. The yi represents the type of the i-th training vector. αi is coefficient which can be solved by quadratic programming. The b is the intercept parameter.  is a kernel function which defines an inner product in a high dimensional feature space. Because of its effectiveness and speed in nonlinear classification process, the radial basis kernel function (RBF)
is a kernel function which defines an inner product in a high dimensional feature space. Because of its effectiveness and speed in nonlinear classification process, the radial basis kernel function (RBF)  was used to in this work.
was used to in this work.2.5. Prediction Assessment




3. Experimental

| Family | This Paper | SVM [ 5] | Naïve Bayes [ 5] | Random Forest [ 5] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sn (%) | Sp (%) | MCC | Sn (%) | Sp (%) | MCC | Sn (%) | Sp (%) | MCC | Sn (%) | Sp (%) | MCC | |
| Kv1 | 100.00 | 96.30 | 0.95 | 93.90 | 93.98 | 0.86 | 93.90 | 83.85 | 0.76 | 97.56 | 78.51 | 0.76 |
| Kv2 | 93.75 | 100.00 | 0.96 | 87.50 | 98.95 | 0.86 | 81.25 | 100.00 | 0.89 | 75.00 | 98.78 | 0.82 |
| Kv3 | 97.30 | 98.89 | 0.95 | 89.19 | 97.69 | 0.87 | 81.08 | 95.12 | 0.75 | 59.45 | 97.44 | 0.67 |
| Kv4 | 100.00 | 100.00 | 1.00 | 93.75 | 100.00 | 0.96 | 87.50 | 100.00 | 0.92 | 65.38 | 98.73 | 0.75 |
| Kv6 | 80.00 | 100.00 | 0.89 | 100.00 | 100.00 | 1.00 | 40.00 | 100.00 | 0.62 | 80.00 | 98.82 | 0.87 |
| Kv7 | 92.50 | 100.00 | 0.95 | 95.00 | 99.39 | 0.95 | 85.00 | 98.70 | 0.87 | 85.00 | 99.29 | 0.89 |
| Average Sn (%) | 93.92 | 93.22 | 78.12 | 77.07 | ||||||||
| Average Sp (%) | 99.20 | 98.34 | 96.28 | 95.26 | ||||||||
| OA (%) | 96.77 | 93.09 | 85.71 | 82.03 | ||||||||
| Method | Sn (%) | OA (%) | |||||
|---|---|---|---|---|---|---|---|
| Kv1 | Kv2 | Kv3 | Kv4 | Kv6 | Kv7 | ||
| Optimal tripeptides (Our method) | 100.00 | 93.75 | 97.30 | 100.00 | 80.00 | 92.50 | 96.77 |
| Optimal tripeptides (SVM-RFE) | 100.00 | 81.25 | 91.67 | 96.88 | 80.00 | 87.55 | 93.09 |
| Traditional PseAAC | 82.93 | 81.25 | 72.97 | 78.13 | 80.00 | 87.50 | 81.11 |
| Optimal tripeptides (Our method) + PseAAC | 100.00 | 87.50 | 97.30 | 100.00 | 80.00 | 92.50 | 96.31 |
| Optimal tripeptides (Our method) + Dipeptides | 100.00 | 81.25 | 94.59 | 100.00 | 80.00 | 92.50 | 95.39 |
4. Conclusions
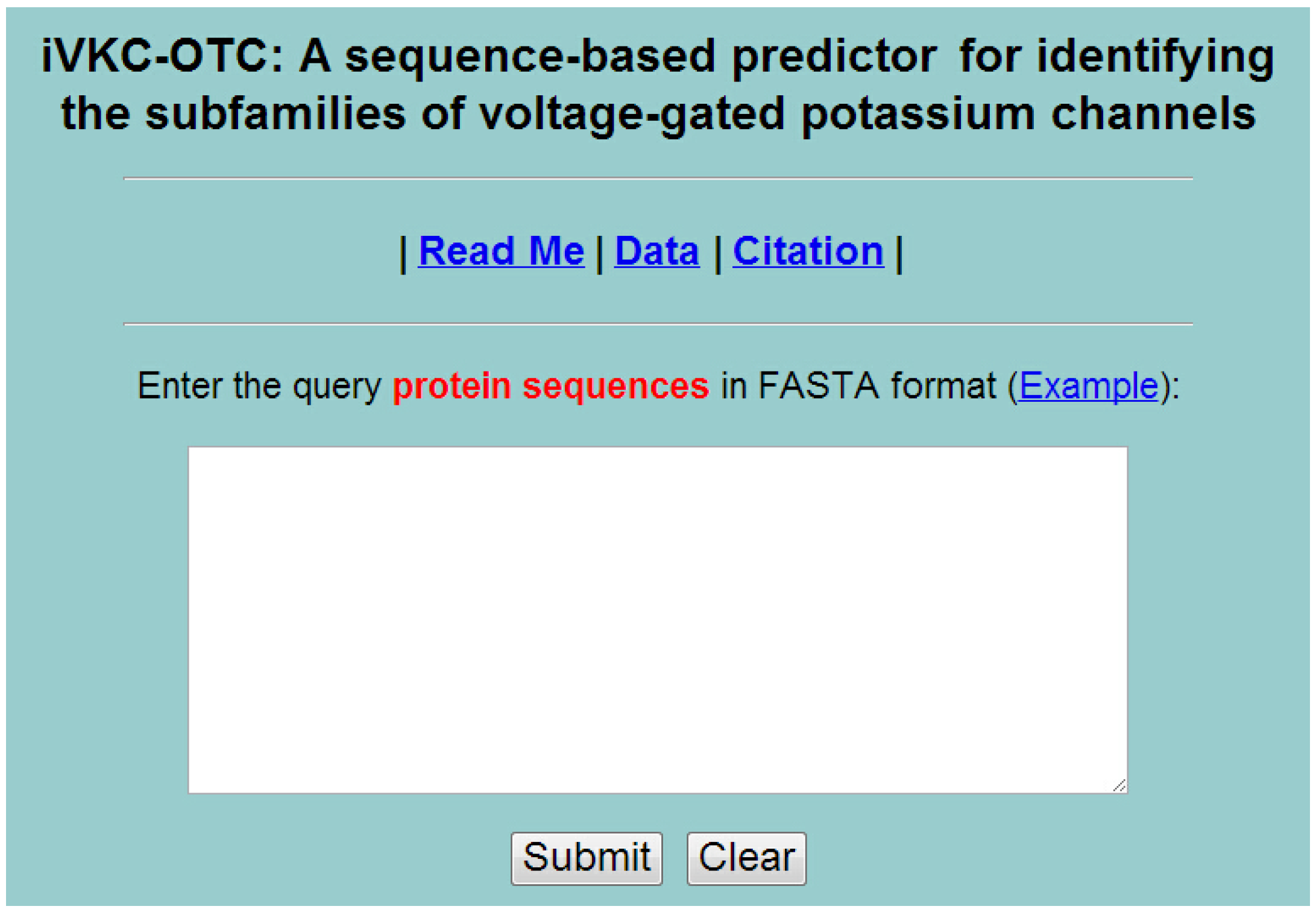
5. Web-Server and User Guide

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Littleton, J.T.; Ganetzky, B. Ion channels and synaptic organization: Analysis of the Drosophila genome. Neuron 2000, 26, 35–43. [Google Scholar] [CrossRef]
- Gallin, W.J.; Boutet, P.A. VKCDB: Voltage-gated K+ channel database updated and upgraded. Nucleic Acids Res. 2011, 39, D362–D366. [Google Scholar] [CrossRef]
- Lehmann-Horn, F.; Jurkat-Rott, K. Voltage-gated ion channels and hereditary disease. Physiol. Rev. 1999, 79, 1317–1372. [Google Scholar]
- Liu, L.X.; Li, M.L.; Tan, F.Y.; Lu, M.C.; Wang, K.L.; Guo, Y.Z.; Wen, Z.N.; Jiang, L. Local sequence information-based support vector machine to classify voltage-gated potassium channels. Acta Biochim. Biophys. Sin. 2006, 38, 363–371. [Google Scholar] [CrossRef]
- Chen, W.; Lin, H. Identification of voltage-gated potassium channel subfamilies from sequence information using support vector machine. Comput. Biol. Med. 2012, 42, 504–507. [Google Scholar] [CrossRef]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Anishetty, S.; Pennathur, G.; Anishetty, R. Tripeptide analysis of protein structures. BMC Struct. Biol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Ung, P.; Winkler, D.A. Tripeptide motifs in biology: Targets for peptidomimetic design. J. Med. Chem. 2011, 54, 1111–1125. [Google Scholar] [CrossRef]
- Ma, J.; Gu, H. A novel method for predicting protein subcellular localization based on pseudo amino acid composition. BMB Rep. 2010, 43, 670–676. [Google Scholar] [CrossRef]
- Olivier, I.; Loots du, T. A metabolomics approach to characterise and identify various Mycobacterium species. J. Microbiol. Methods 2012, 88, 419–426. [Google Scholar] [CrossRef]
- Yin, J.B.; Fan, Y.X.; Shen, H.B. Conotoxin superfamily prediction using diffusion maps dimensionality reduction and subspace classifier. Curr. Protein Pept. Sci. 2011, 12, 580–588. [Google Scholar] [CrossRef]
- Jia, P.; Qian, Z.; Feng, K.; Lu, W.; Li, Y.; Cai, Y. Prediction of membrane protein types in a hybrid space. J. Proteome Res. 2008, 7, 1131–1137. [Google Scholar] [CrossRef]
- Huang, T.; Xu, Z.; Chen, L.; Cai, Y.D.; Kong, X. Computational analysis of HIV-1 resistance based on gene expression profiles and the virus-host interaction network. PLoS One 2011, 6, e17291. [Google Scholar]
- Rashid, M.; Saha, S.; Raghava, G.P. Support vector machine-based method for predicting subcellular localization of mycobacterial proteins using evolutionary information and motifs. BMC Bioinform. 2007. [Google Scholar] [CrossRef]
- Liu, B.; Xu, J.; Zou, Q.; Xu, R.; Wang, X.; Chen, Q. Using distances between top-n-gram and residue pairs for protein remote homology detection. BMC Bioinform. 2014. [Google Scholar] [CrossRef]
- Liu, B.; Wang, X.; Lin, L.; Tang, B.; Dong, Q.; Wang, X. Prediction of protein binding sites in protein structures using hidden Markov support vector machine. BMC Bioinform. 2009. [Google Scholar] [CrossRef]
- Liu, B.; Wang, X.; Lin, L.; Dong, Q.; Wang, X. A discriminative method for protein remote homology detection and fold recognition combining Top-n-grams and latent semantic analysis. BMC bioinform. 2008. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, D.; Xu, R.; Xu, J.; Wang, X.; Chen, Q.; Dong, Q.; Chou, K.C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics 2013, 30, 472–479. [Google Scholar]
- Liu, B.; Xu, J.; Fan, S.; Xu, R.; Zhou, J.; Wang, X. Protein remote homology detection by combining Chou’s pseudo amino acid composition and profile—Based protein representation. Mol. Inform. 2013, 32, 775–782. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Chou, K.C.; Zhang, C.T. Prediction of protein structural classes. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 275–349. [Google Scholar] [CrossRef]
- Lin, H.; Chen, W.; Yuan, L.F.; Li, Z.Q.; Ding, H. Using over-represented tetrapeptides to predict protein submitochondria locations. Acta Biotheor. 2013, 61, 259–268. [Google Scholar] [CrossRef]
- Fan, G.L.; Liu, Y.L.; Zuo, Y.C.; Mei, H.X.; Rang, Y.; Hou, B.Y.; Zhao, Y. acACS: Improving the prediction accuracy of protein subcellular locations and protein classification by incorporating the average chemical shifts composition. Sci. World J. 2014. [Google Scholar] [CrossRef]
- Lin, H.; Ding, H.; Guo, F.B.; Huang, J. Prediction of subcellular location of mycobacterial protein using feature selection techniques. Mol. Divers. 2010, 14, 667–671. [Google Scholar] [CrossRef]
- Li, L.; Yu, S.; Xiao, W.; Li, Y.; Li, M.; Huang, L.; Zheng, X.; Zhou, S.; Yang, H. Prediction of bacterial protein subcellular localization by incorporating various features into Chou’s PseAAC and a backward feature selection approach. Biochimie 2014. [Google Scholar] [CrossRef]
- Li, L.; Cui, X.; Yu, S.; Zhang, Y.; Luo, Z.; Yang, H.; Zhou, Y.; Zheng, X. PSSP-RFE: Accurate prediction of protein structural class by recursive feature extraction from psi-blast profile, physical-chemical property and functional annotations. PLoS One 2014, 9, e92863. [Google Scholar]
- The Webserver iVKC-OTC. Available online: http://lin.uestc.edu.cn/server/iVKC-OTC (accessed on 14 July 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, W.-X.; Deng, E.-Z.; Chen, W.; Lin, H. Identifying the Subfamilies of Voltage-Gated Potassium Channels Using Feature Selection Technique. Int. J. Mol. Sci. 2014, 15, 12940-12951. https://doi.org/10.3390/ijms150712940
Liu W-X, Deng E-Z, Chen W, Lin H. Identifying the Subfamilies of Voltage-Gated Potassium Channels Using Feature Selection Technique. International Journal of Molecular Sciences. 2014; 15(7):12940-12951. https://doi.org/10.3390/ijms150712940
Chicago/Turabian StyleLiu, Wei-Xin, En-Ze Deng, Wei Chen, and Hao Lin. 2014. "Identifying the Subfamilies of Voltage-Gated Potassium Channels Using Feature Selection Technique" International Journal of Molecular Sciences 15, no. 7: 12940-12951. https://doi.org/10.3390/ijms150712940