BRCA1 Exon 11, a CERES (Composite Regulatory Element of Splicing) Element Involved in Splice Regulation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
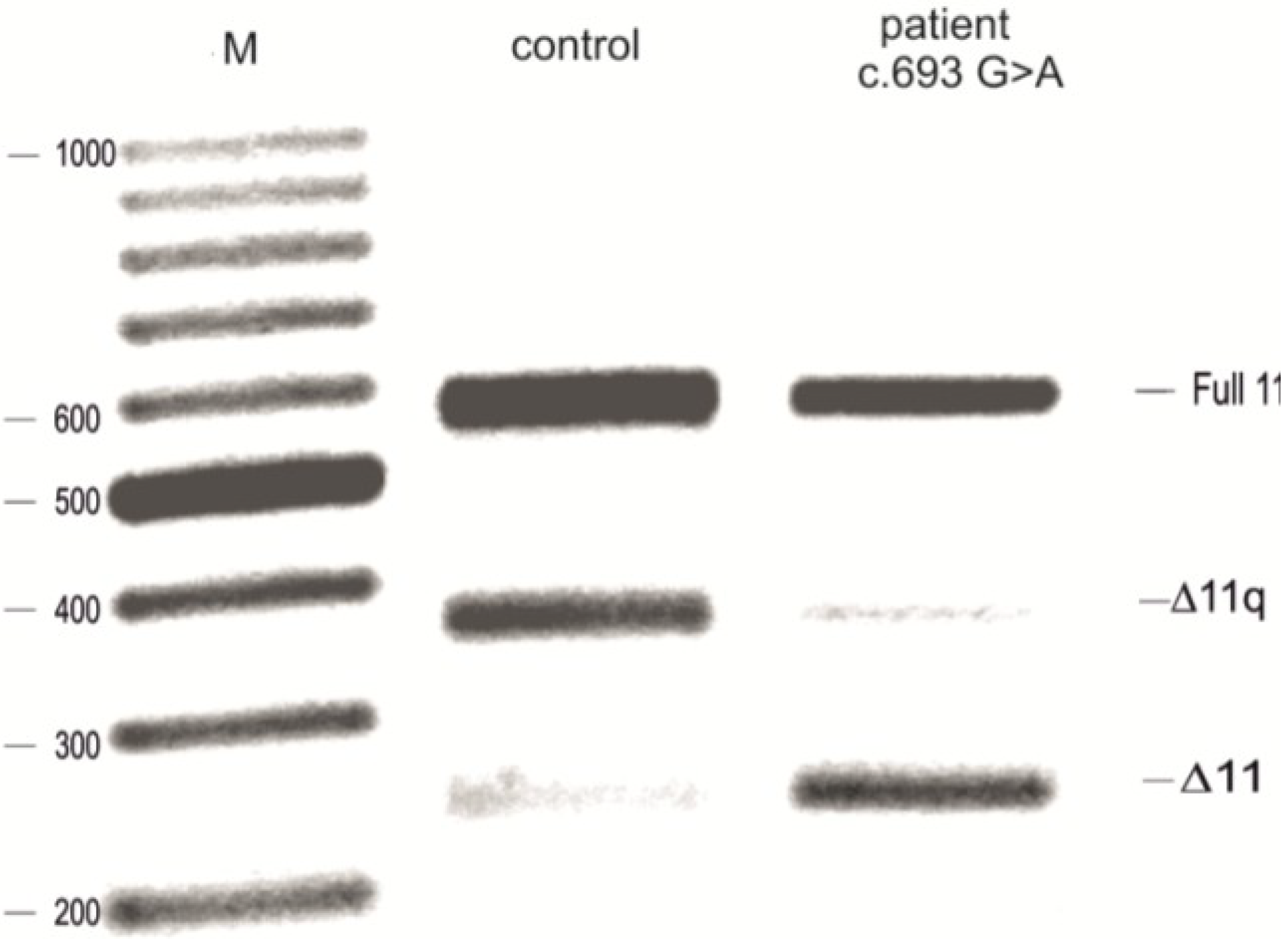
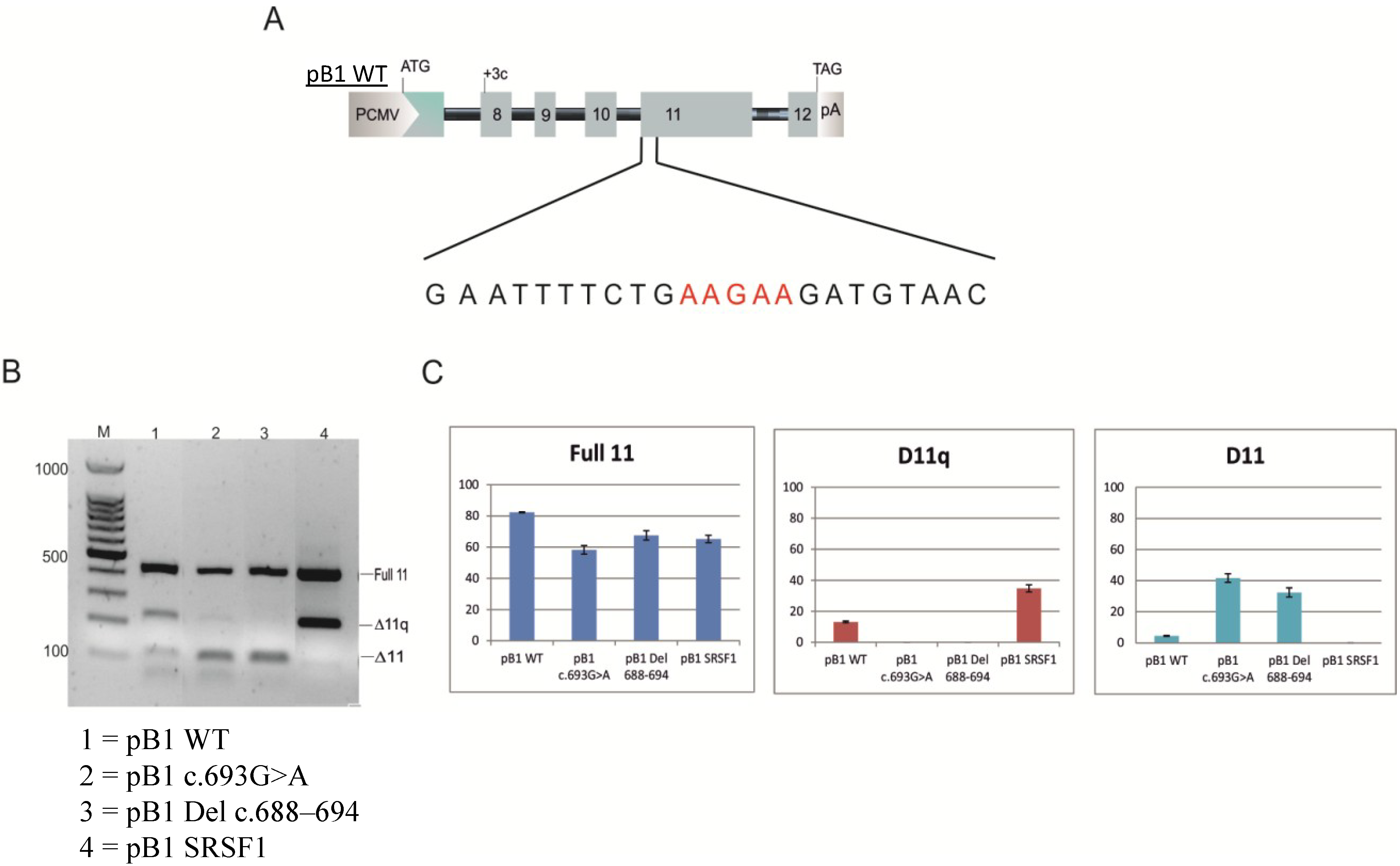
2.1. The UV (Unclassified Variant) c.693G>A Affects Splicing of Exon 11

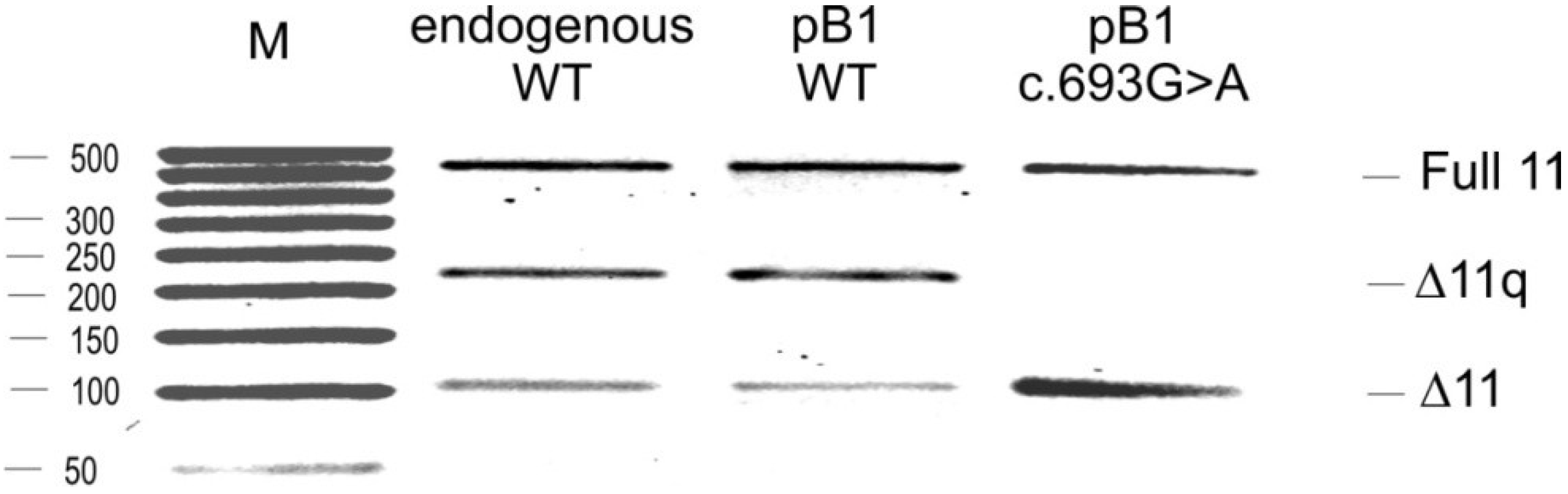
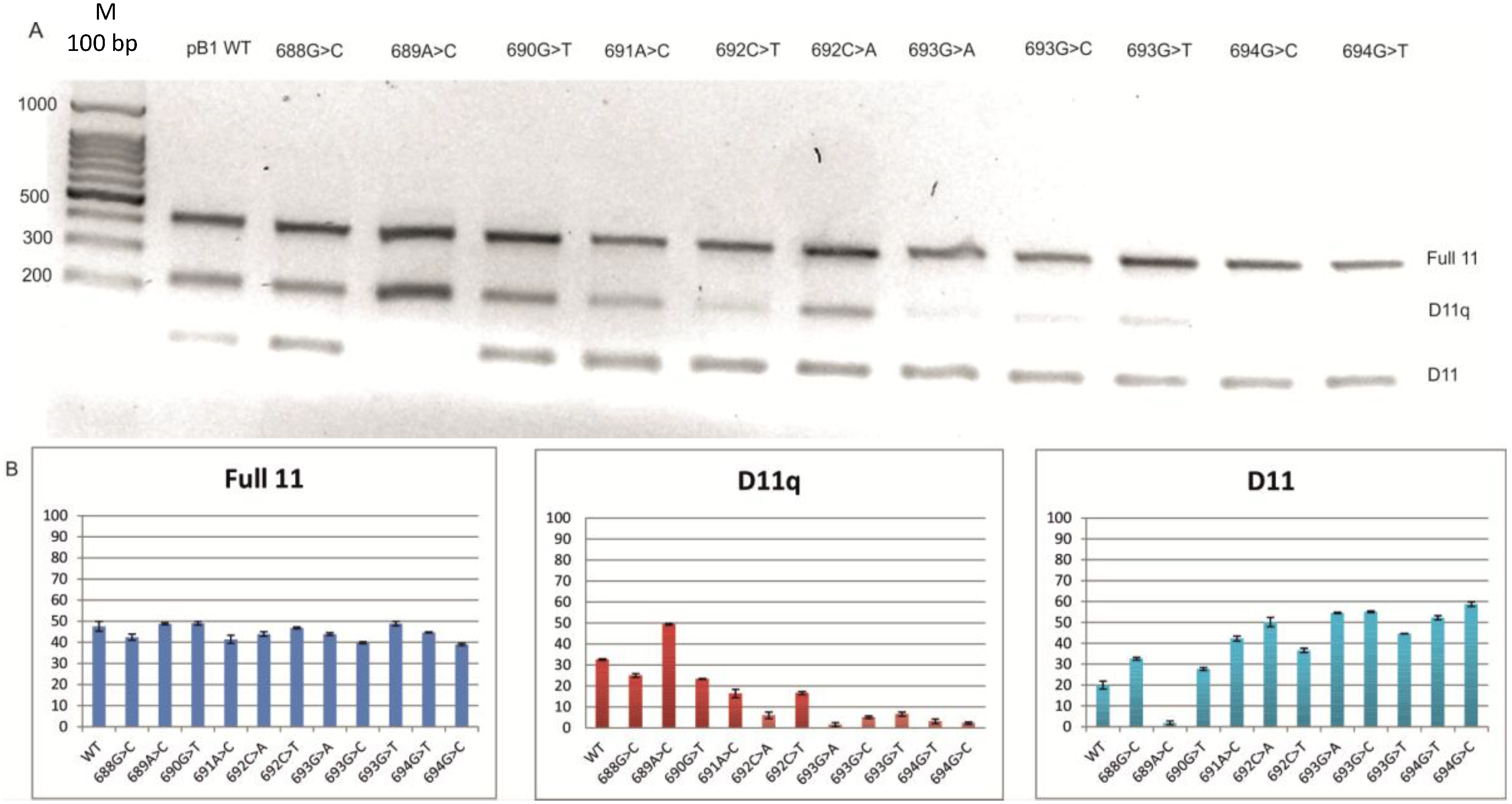
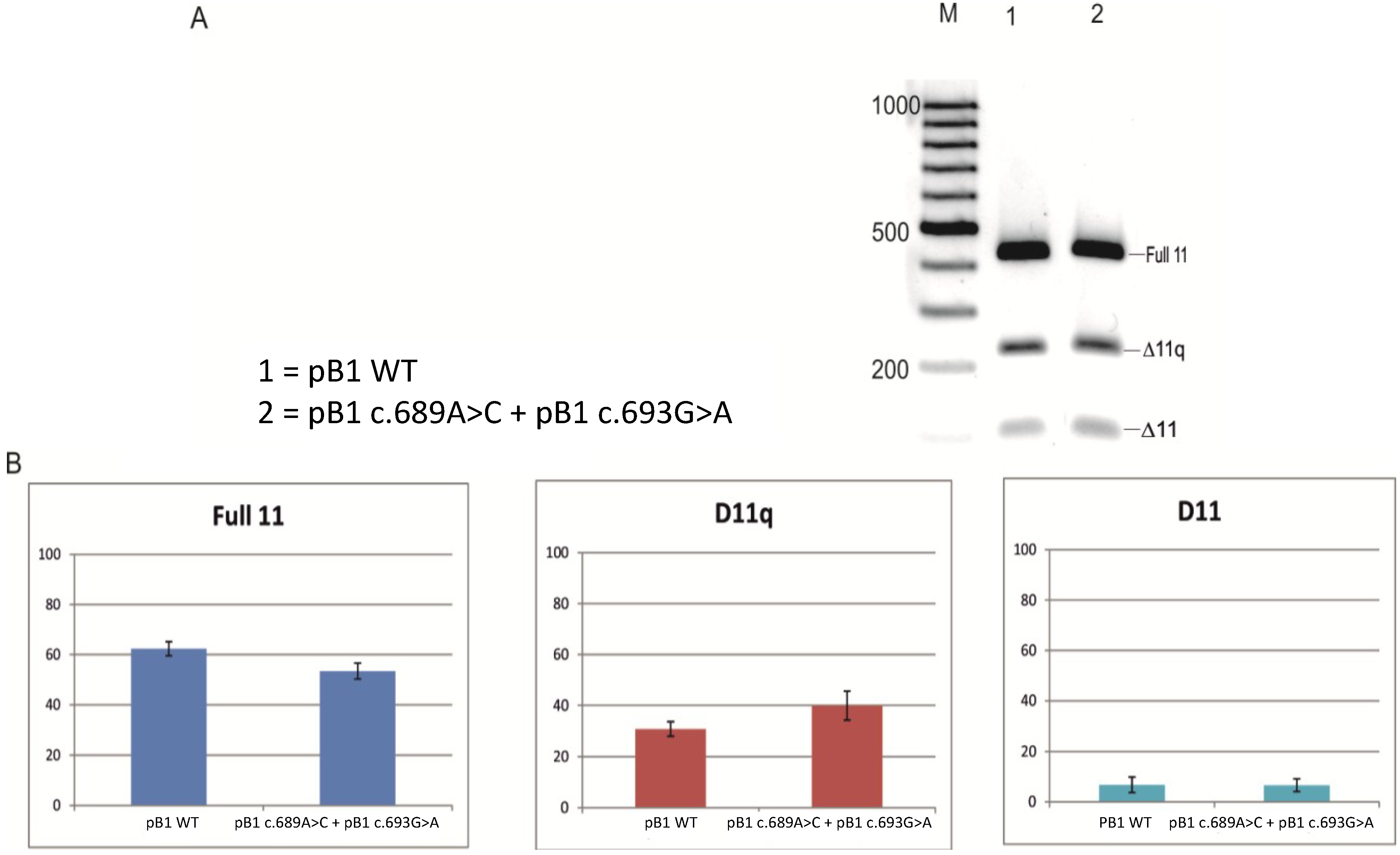
2.2. Minigene Analysis





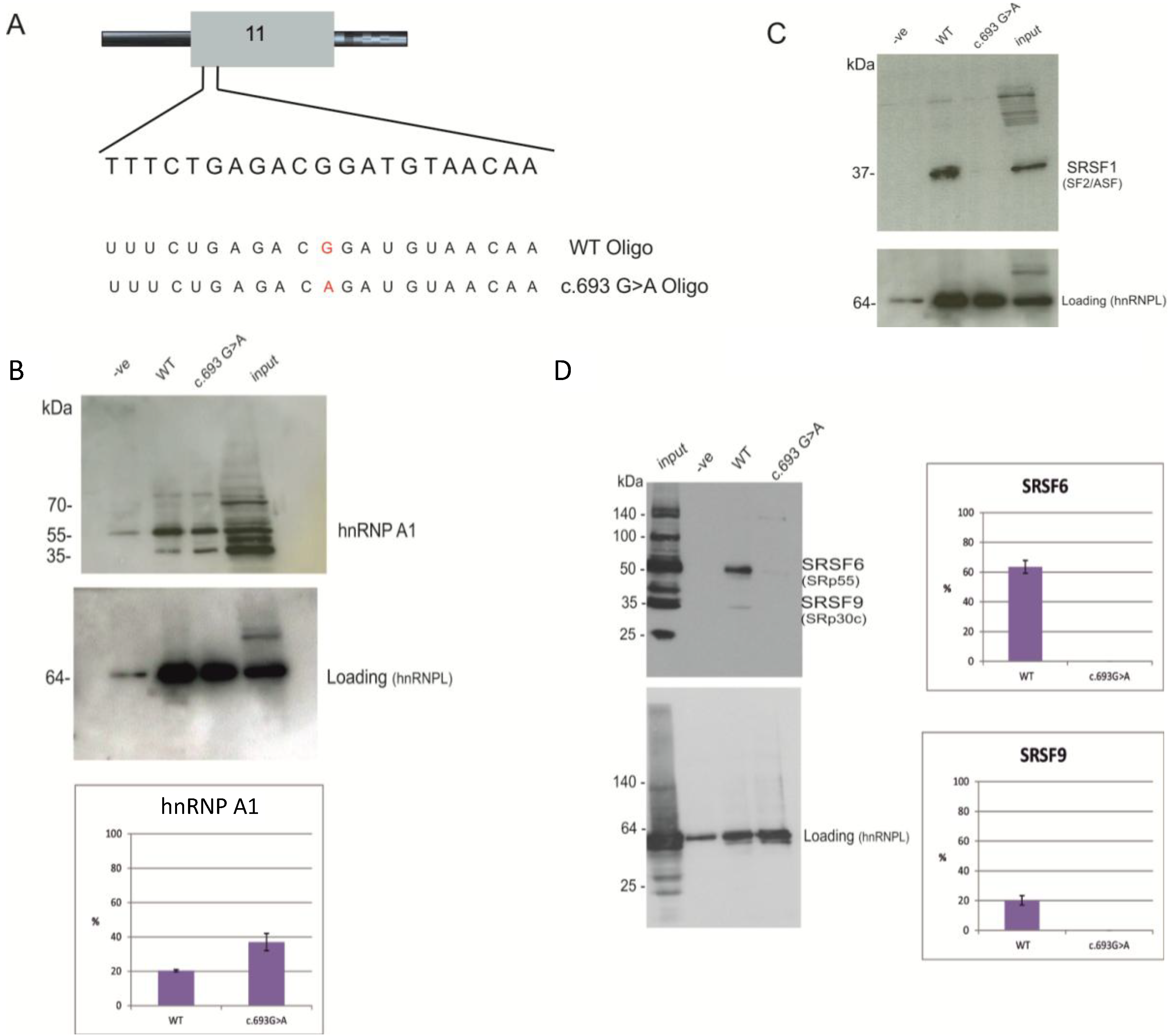
2.3. Identification of Proteins Binding to the Splicing Regulatory Element of BRCA1 Exon 11


3. Experimental Section
3.1. Minigene Constructs
3.2. Cell Culture
3.3. Transfection
3.4. Pull-Down Assay
3.5. Western Blot
3.6. In Silico Predictions
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Staley, J.P.; Guthrie, C. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell 1998, 92, 315–326. [Google Scholar] [CrossRef]
- Lamond, A.I. The spliceosome. Bioessays 1993, 15, 595–603. [Google Scholar] [CrossRef]
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef]
- Orban, T.I.; Olah, E. Expression profiles of BRCA1 splice variants in asynchronous and in G1/S synchronized tumor cell lines. Biochem. Biophys. Res. Commun. 2001, 280, 32–38. [Google Scholar] [CrossRef]
- Orban, T.I.; Olah, E. Emerging roles of BRCA1 alternative splicing. Mol. Pathol. 2003, 56, 191–197. [Google Scholar] [CrossRef]
- Xu, C.F.; Chambers, J.A.; Nicolai, H.; Brown, M.A.; Hujeirat, Y.; Mohammed, S.; Hodgson, S.; Kelsell, D.P.; Spurr, N.K.; Bishop, D.T.; et al. Mutations and alternative splicing of the BRCA1 gene in UK breast/ovarian cancer families. Genes Chromosom. Cancer 1997, 18, 102–110. [Google Scholar]
- Wilson, C.A.; Payton, M.N.; Elliott, G.S.; Buaas, F.W.; Cajulis, E.E.; Grosshans, D.; Ramos, L.; Reese, D.M.; Slamon, D.J.; Calzone, F.J. Differential subcellular localization, expression and biological toxicity of BRCA1 and the splice variant BRCA1-delta11b. Oncogene 1997, 14, 1–16. [Google Scholar]
- Miao, L.; Cao, Z.; Shen, C.; Gu, C.; Zheng, C. Alternative splicing of breast cancer associated gene BRCA1 from breast cancer cell line. J. Biochem. Mol. Biol. 2007, 40, 15–21. [Google Scholar] [CrossRef]
- Tammaro, C.; Raponi, M.; Wilson, D.I.; Baralle, D. BRCA1 exon 11 alternative splicing, multiple functions and the association with cancer. Biochem. Soc. Trans. 2012, 40, 768–772. [Google Scholar] [CrossRef]
- Huber, L.J.; Yang, T.W.; Sarkisian, C.J.; Master, S.R.; Deng, C.-X.; Chodosh, L. A Impaired DNA damage response in cells expressing an exon 11-deleted murine brca1 variant that localizes to nuclear foci. Mol. Cell. Biol. 2001, 21, 4005–4015. [Google Scholar] [CrossRef]
- Raponi, M.; Baralle, D. Alternative splicing: good and bad effects of translationally silent substitutions. FEBS J. 2010, 277, 836–840. [Google Scholar] [CrossRef]
- Pagani, F.; Raponi, M.; Baralle, F.E. Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc. Natl. Acad. Sci. USA 2005, 102, 6368–6372. [Google Scholar] [CrossRef]
- Brandao, R.D.; van Roozendaal, K.; Tserpelis, D.; Garcia, E.G.; Blok, M.J. Characterisation of unclassified variants in the BRCA1/2 genes with a putative effect on splicing. Breast Cancer Res. Treat. 2011, 129, 971–982. [Google Scholar] [CrossRef]
- Raponi, M.; Douglas, A.G.; Tammaro, C.; Wilson, D.I.; Baralle, D. Evolutionary constraint helps unmask a splicing regulatory region in BRCA1 exon 11. PLoS One 2012, 7, e37255. [Google Scholar]
- Pagani, F.; Stuani, C.; Tzetis, M.; Kanavakis, E.; Efthymiadou, A.; Doudounakis, S.; Casals, T.; Baralle, F.E. New type of disease causing mutations: the example of the composite exonic regulatory elements of splicing in CFTR exon 12. Hum. Mol. Genet. 2003, 12, 1111–1120. [Google Scholar] [CrossRef]
- Pagani, F.; Buratti, E.; Stuani, C.; Baralle, F.E. Missense, nonsense, and neutral mutations define juxtaposed regulatory elements of splicing in cystic fibrosis transmembrane regulator exon 9. J. Biol. Chem. 2003, 278, 26580–26588. [Google Scholar]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37. [Google Scholar] [CrossRef]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder Version 2.4.1. Available online: http://www.umd.be/HSF/ (accessed on 19 January 2014).
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.F.; Revil, T.; Chabot, B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007, 623, 123–147. [Google Scholar] [CrossRef]
- Park, E.; Han, J.; Son, G.H.; Lee, M.S.; Chung, S.; Park, S.H.; Park, K.; Lee, K.H.; Choi, S.; Seong, J.Y.; et al. Cooperative actions of Tra2α with 9G8 and SRp30c in the RNA splicing of the gonadotropin-releasing hormone gene transcript. J. Biol. Chem. 2006, 281, 401–409. [Google Scholar] [CrossRef]
- Good, L.; Nazar, R.N. An improved thermal cycle for two-step PCR-based targeted mutagenesis. Nucleic Acids Res. 1992, 20, 4934. [Google Scholar] [CrossRef]
- Kim, S.S.; Cao, L.; Lim, S.C.; Li, C.; Wang, R.H.; Xu, X.; Bachelier, R.; Deng, C.X. Hyperplasia and spontaneous tumor development in the gynecologic system in mice lacking the BRCA1-Δ11 isoform. Mol. Cell Biol. 2006, 26, 6983–6992. [Google Scholar] [CrossRef]
- Xu, X.L.; Qiao, W.H.; Linke, S.P.; Cao, L.; Li, W.M.; Furth, P.A.; Harris, C.C.; Deng, C.X. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat. Genet. 2001, 28, 266–271. [Google Scholar] [CrossRef]
- Fabbro, M.; Rodriguez, J.A.; Baer, R.; Henderson, B.R. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J. Biol. Chem. 2002, 277, 21315–21324. [Google Scholar]
- Qin, Y.; Xu, J.; Aysola, K.; Begum, N.; Reddy, V.; Chai, Y.; Grizzle, W.E.; Partridge, E.E.; Reddy, E.S.P.; Rao, V.N. Ubc9 mediates nuclear localization and growth suppression of BRCA1 and BRCA1a proteins. J. Cell. Physiol. 2011, 226, 3355–3367. [Google Scholar] [CrossRef]
- Raponi, M.; Kralovicova, J.; Copson, E.; Divina, P.; Eccles, D.; Johnson, P.; Baralle, D.; Vorechovsky, I. Prediction of single-nucleotide substitutions that result in exon skipping: Identification of a splicing silencer in BRCA1 exon 6. Hum. Mutat. 2011, 32, 436–444. [Google Scholar] [CrossRef]
- Raponi, M.; Buratti, E.; Llorian, M.; Stuani, C.; Smith, C.W.J.; Baralle, D. Polypyrimidine tract binding protein regulates alternative splicing of an aberrant pseudoexon in NF1. FEBS J. 2008, 275, 6101–6108. [Google Scholar]
- Raponi, M.; Buratti, E.; Dassie, E.; Upadhyaya, M.; Baralle, D. Low U1 snRNP dependence at the NF1 exon 29 donor splice site. FEBS J. 2009, 276, 2060–2073. [Google Scholar]
- Haque, A.; Buratti, E.; Baralle, F.E. Functional properties and evolutionary splicing constraints on a composite exonic regulatory element of splicing in CFTR exon 12. Nucleic Acids Res. 2010, 38, 647–659. [Google Scholar] [CrossRef]
- Cartegni, L.; Hastings, M.L.; Calarco, J.A.; de Stanchina, E.; Krainer, A.R. Determinants of exon 7 splicing in the spinal muscular atrophy genes, SMN1 and SMN2. Am. J. Hum. Genet. 2006, 78, 63–77. [Google Scholar] [CrossRef]
- Singh, N.N.; Androphy, E.J.; Singh, R.N. An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy. Biochem. Biophys. Res. Commun. 2004, 315, 381–388. [Google Scholar] [CrossRef]
- Kashima, T.; Rao, N.; David, C.J.; Manley, J.L. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum. Mol. Genet. 2007, 16, 3149–3159. [Google Scholar] [CrossRef]
- Krainer, A.R.; Conway, G.C.; Kozak, D. The essential pre-mRNA splicing factor SF2 influences 5' splice site selection by activating proximal sites. Cell 1990, 62, 35–42. [Google Scholar] [CrossRef]
- Ge, H.; Manley, J.L. A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell 1990, 62, 25–34. [Google Scholar] [CrossRef]
- Jensen, M.A.; Wilkinson, J.E.; Krainer, A.R. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat. Struct. Mol. Biol. 2014, 21, 189–197. [Google Scholar] [CrossRef]
- Simard, M.J.; Chabot, B. SRp30c is a repressor of 3' splice site utilization. Mol. Cell. Biol. 2002, 22, 4001–4010. [Google Scholar] [CrossRef]
- Whiley, P.J.; de la Hoya, M.; Thomassen, M.; Becker, A.; Brandao, R.; Pedersen, I.S.; Montagna, M.; Menendez, M.; Quiles, F.; Gutierrez-Enriquez, S.; et al. Comparison of mRNA splicing assay protocols across multiple laboratories: Recommendations for best practice in standardized clinical testing. Clin. Chem. 2014, 60, 341–352. [Google Scholar] [CrossRef] [Green Version]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tammaro, C.; Raponi, M.; Wilson, D.I.; Baralle, D. BRCA1 Exon 11, a CERES (Composite Regulatory Element of Splicing) Element Involved in Splice Regulation. Int. J. Mol. Sci. 2014, 15, 13045-13059. https://doi.org/10.3390/ijms150713045
Tammaro C, Raponi M, Wilson DI, Baralle D. BRCA1 Exon 11, a CERES (Composite Regulatory Element of Splicing) Element Involved in Splice Regulation. International Journal of Molecular Sciences. 2014; 15(7):13045-13059. https://doi.org/10.3390/ijms150713045
Chicago/Turabian StyleTammaro, Claudia, Michela Raponi, David I. Wilson, and Diana Baralle. 2014. "BRCA1 Exon 11, a CERES (Composite Regulatory Element of Splicing) Element Involved in Splice Regulation" International Journal of Molecular Sciences 15, no. 7: 13045-13059. https://doi.org/10.3390/ijms150713045