DNA Break Mapping Reveals Topoisomerase II Activity Genome-Wide

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
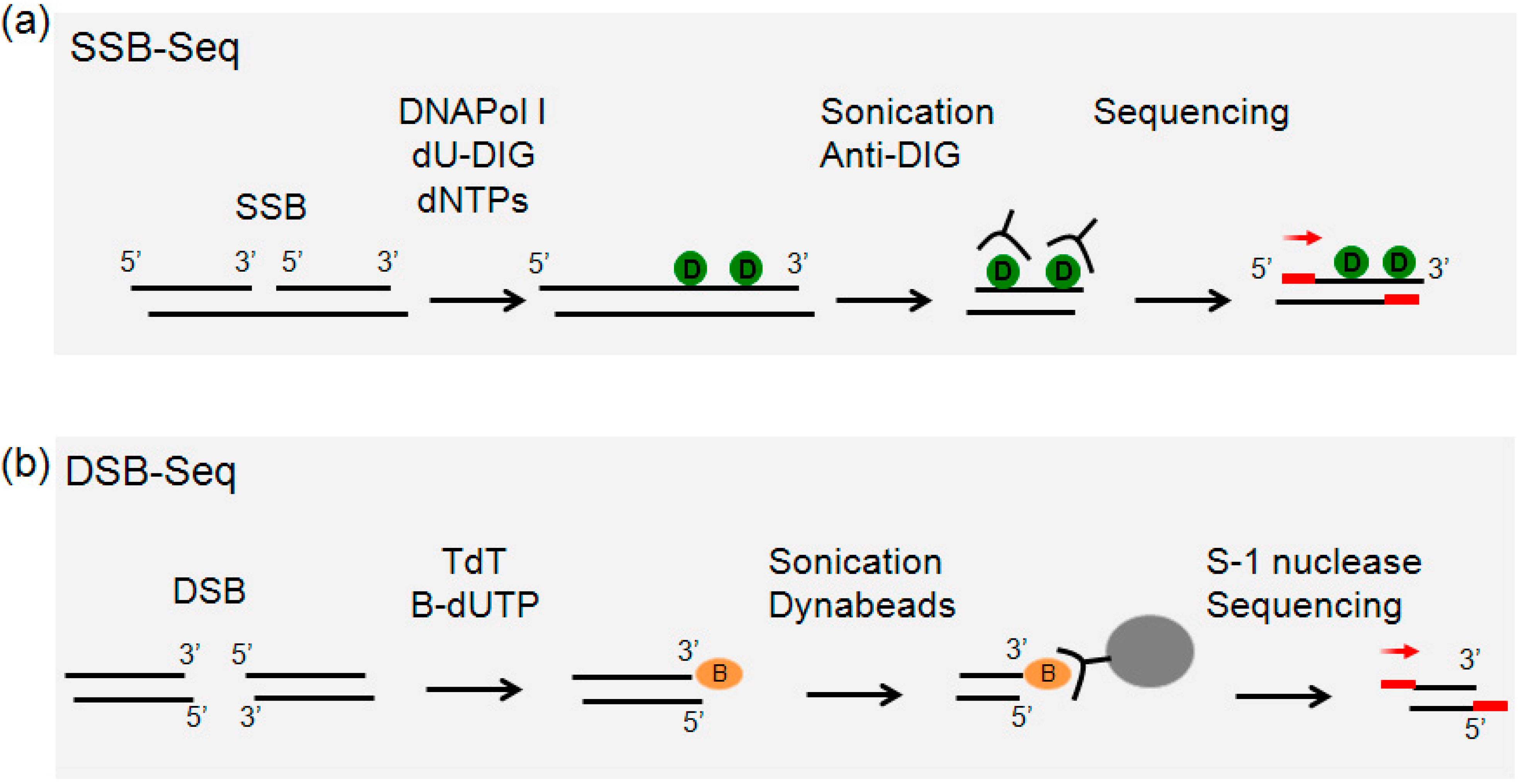
2.1. Overview of Single-Strand Break (SSB)-Seq

2.2. Overview of Double-Strand Break (DSB)-Seq
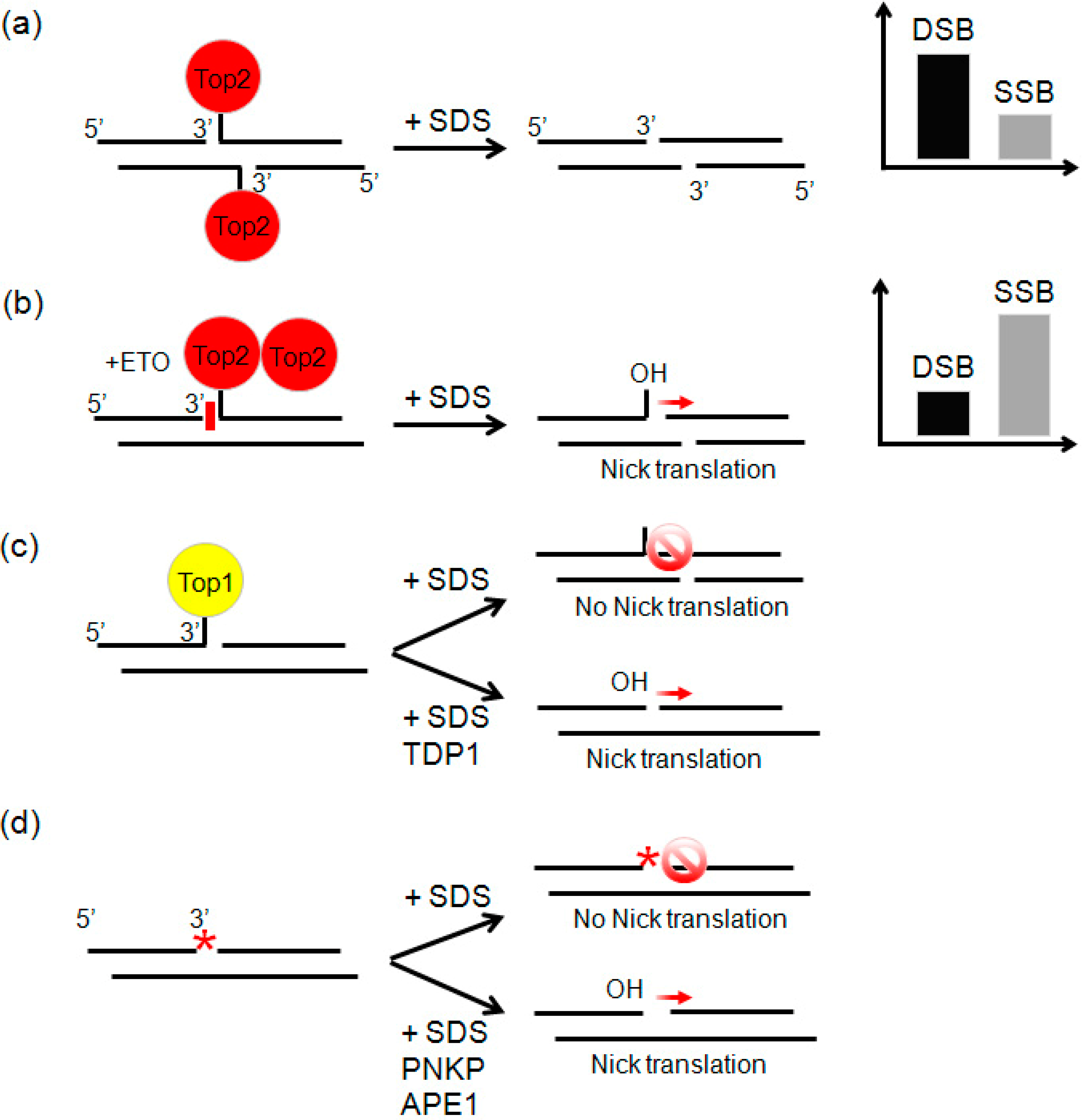
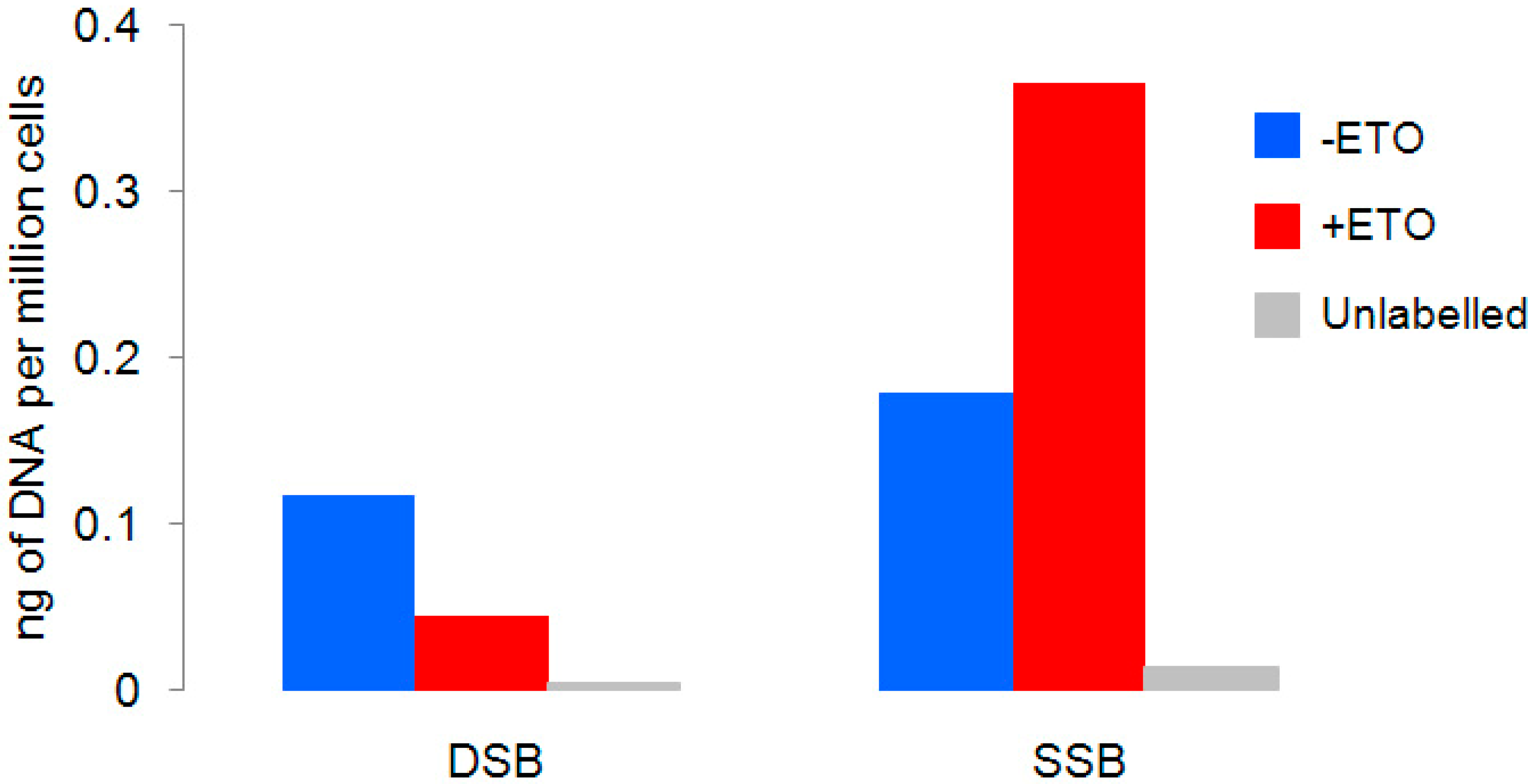
2.3. Etoposide Induces SSBs
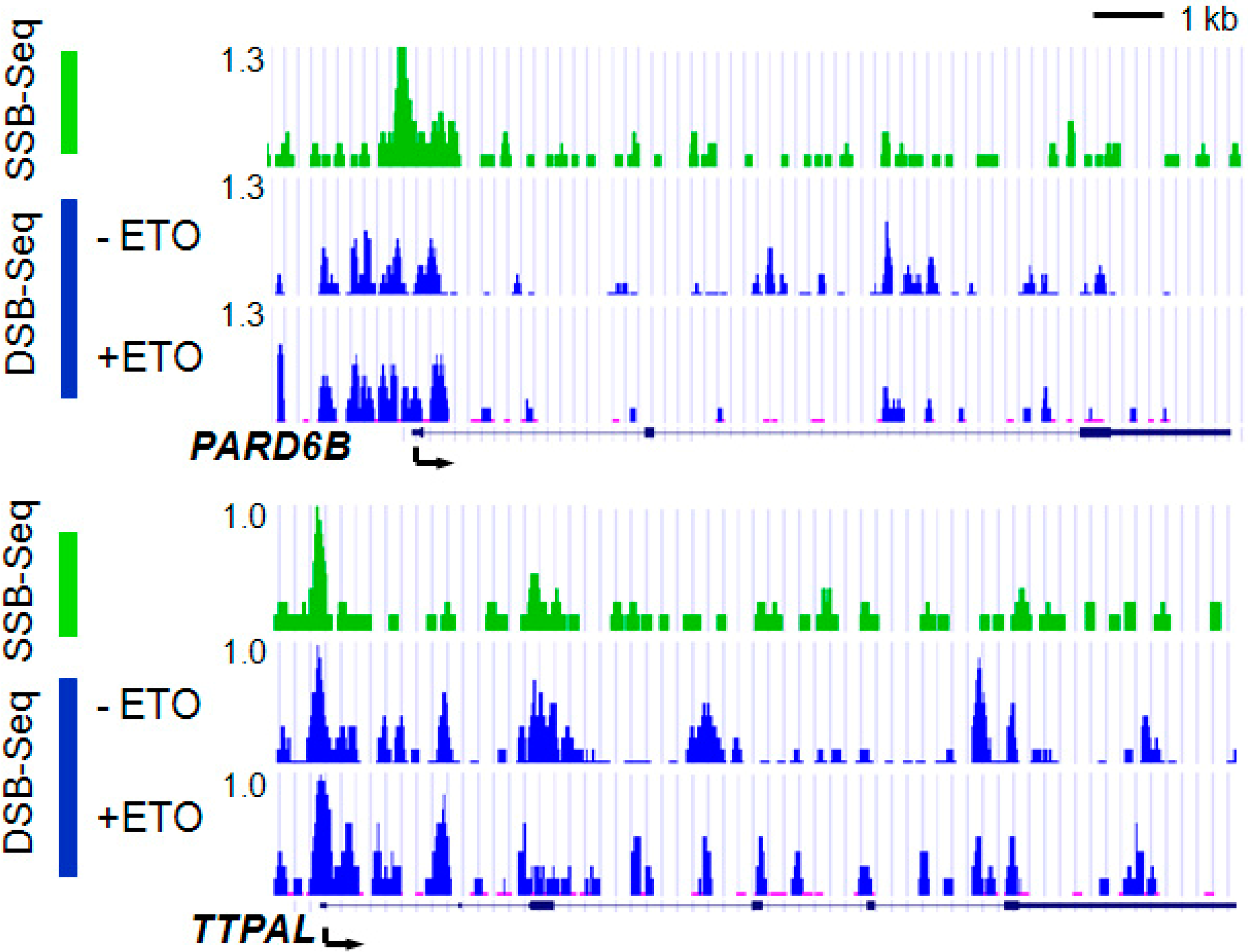
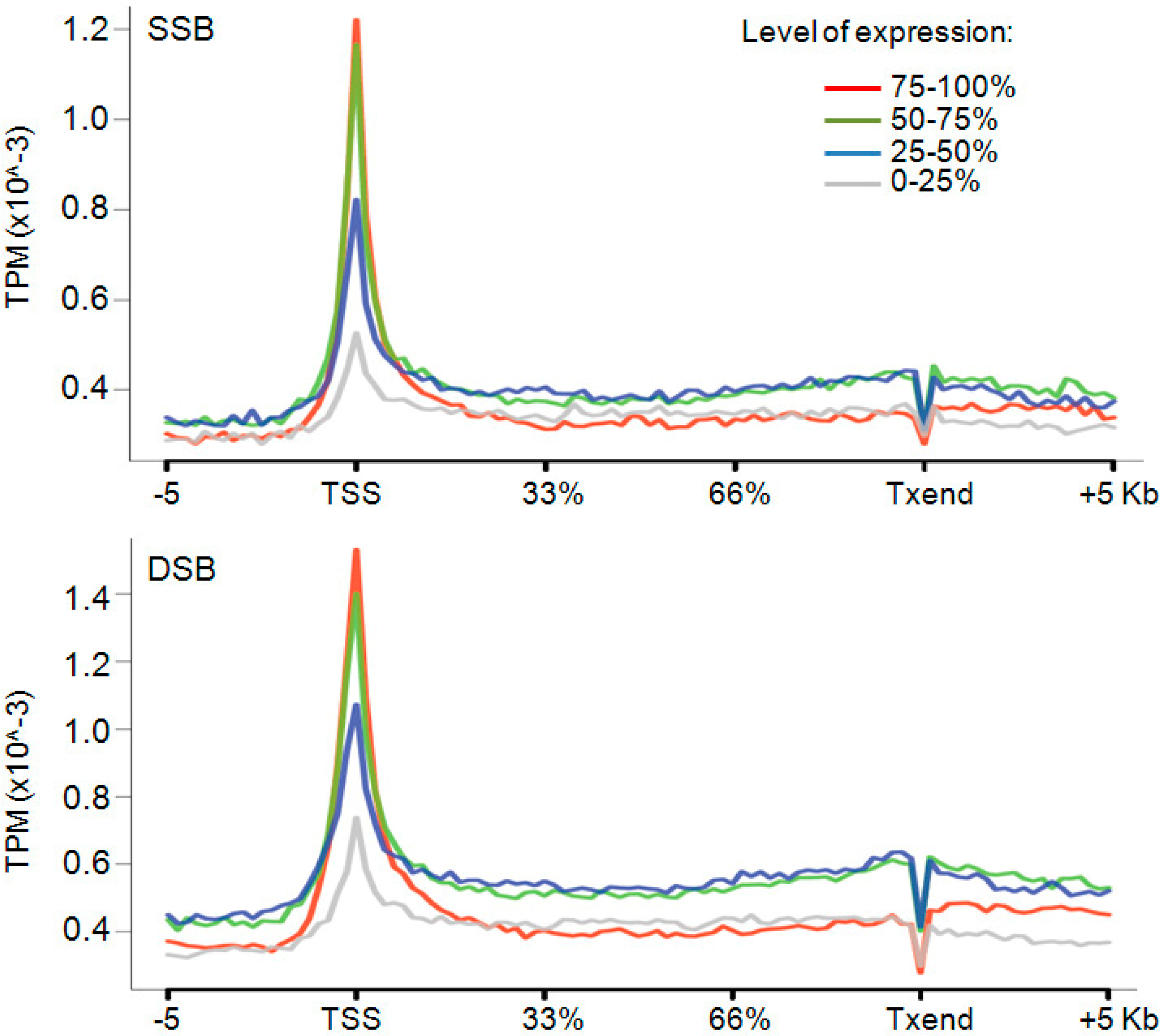
2.4. Promoters Are Hot Spots of DNA Breaks




3. Experimental Section
3.1. Cells and Reagents
3.2. Purification of High Molecular Weight DNA
3.3. SSB-Seq
3.4. DSB-Seq
3.5. Template Preparation for Sequencing Analysis
3.6. Data Analysis of Microarrays
3.7. Processing of Sequencing Data
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Aguilera, A.; Garcia-Muse, T. Causes of genome instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef]
- Bennett, C.B.; Lewis, A.L.; Baldwin, K.K.; Resnick, M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 1993, 90, 5613–5617. [Google Scholar] [CrossRef]
- Sandell, L.L.; Zakian, V.A. Loss of a yeast telomere: Arrest, recovery, and chromosome loss. Cell 1993, 75, 729–739. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Caldecott, K.W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair 2007, 6, 443–453. [Google Scholar] [CrossRef]
- Ju, B.G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A Topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-resolution profiling of γH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef]
- Blitzblau, H.G.; Hochwagen, A. Genome-wide detection of meiotic DNA double-strand break hotspots using single-stranded DNA. Methods Mol. Biol. 2011, 745, 47–63. [Google Scholar] [CrossRef]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA Topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Baranello, L.; Kouzine, F.; Levens, D. DNA topoisomerases: Beyond the standard role. Transcription 2013, 4, 232–237. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Osheroff, N. Etoposide, Topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar]
- Muslimovic, A.; Nyström, S.; Gao, Y.; Hammarsten, O. Numerical analysis of etoposide induced DNA breaks. PLoS One 2009, 4, e5859. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar]
- Nitiss, J.L. DNA Topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef]
- Miao, Z.H.; Player, A.; Shankavaram, U.; Wang, Y.H.; Zimonjic, D.B.; Lorenzi, P.L.; Liao, Z.Y.; Liu, H.; Shimura, T.; Zhang, H.L.; et al. Nonclassic functions of human Topoisomerase I: Genome-wide and pharmacologic analyses. Cancer Res. 2007, 67, 8752–8761. [Google Scholar] [CrossRef]
- Kouzine, F.; Gupta, A.; Baranello, L.; Wojtowicz, D.; Ben-Aissa, K.; Liu, J.; Przytycka, T.M.; Levens, D. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol. 2013, 20, 396–403. [Google Scholar] [CrossRef]
- Roca, J.; Wang, J.C. The probabilities of supercoil removal and decatenation by yeast DNA Topoisomerase II. Genes Cells 1996, 1, 17–27. [Google Scholar] [CrossRef]
- Lavelle, C. Forces and torques in the nucleus: Chromatin under mechanical constraints. Biochem. Cell Biol. 2009, 87, 307–322. [Google Scholar] [CrossRef]
- Kouzine, F.; Wojtowicz, D.; Yamane, A.; Resch, W.; Kieffer-Kwon, K.R.; Bandle, R.; Nelson, S.; Nakahashi, H.; Awasthi, P.; Feigenbaum, L.; et al. Global regulation of promoter melting in naive lymphocytes. Cell 2013, 153, 988–999. [Google Scholar] [CrossRef]
- Hubbard, T.J.P.; Aken, B.L.; Ayling, S.; Ballester, B.; Beal, K.; Bragin, E.; Brent, S.; Chen, Y.; Clapham, P.; Clarke, L.; et al. Ensembl 2009. NucleicAcids Res. 2009, 37, D690–D697. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Shen, L.; Shao, N.; Liu, X.; Nestler, E. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 2014. [Google Scholar] [CrossRef]
- Cornarotti, M.; Tinelli, S.; Willmore, E.; Zunino, F.; Fisher, L.M.; Austin, C.A.; Capranico, G. Drug sensitivity and sequence specificity of human recombinant DNA topoisomerases IIα (p170) and IIβ (p180). Mol. Pharmacol. 1996, 50, 1463–1471. [Google Scholar]
- Willmore, E.; Errington, F.; Tilby, M.J.; Austin, C.A. Formation and longevity of idarubicin-induced DNA Topoisomerase II cleavable complexes in K562 human leukaemia cells. Biochem. Pharmacol. 2002, 63, 1807–1815. [Google Scholar] [CrossRef]
- Willmore, E.; Frank, A.J.; Padget, K.; Tilby, M.J.; Austin, C.A. Etoposide targets topoisomerase IIα and IIβ in leukemic cells: Isoform-specific cleavable complexes visualized and quantified in situ by a novel immunofluorescence technique. Mol. Pharmacol. 1998, 54, 78–85. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Baranello, L.; Kouzine, F.; Wojtowicz, D.; Cui, K.; Przytycka, T.M.; Zhao, K.; Levens, D. DNA Break Mapping Reveals Topoisomerase II Activity Genome-Wide. Int. J. Mol. Sci. 2014, 15, 13111-13122. https://doi.org/10.3390/ijms150713111
Baranello L, Kouzine F, Wojtowicz D, Cui K, Przytycka TM, Zhao K, Levens D. DNA Break Mapping Reveals Topoisomerase II Activity Genome-Wide. International Journal of Molecular Sciences. 2014; 15(7):13111-13122. https://doi.org/10.3390/ijms150713111
Chicago/Turabian StyleBaranello, Laura, Fedor Kouzine, Damian Wojtowicz, Kairong Cui, Teresa M. Przytycka, Keji Zhao, and David Levens. 2014. "DNA Break Mapping Reveals Topoisomerase II Activity Genome-Wide" International Journal of Molecular Sciences 15, no. 7: 13111-13122. https://doi.org/10.3390/ijms150713111