Receptor for Advanced Glycation End Products (RAGE) and Its Ligands: Focus on Spinal Cord Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
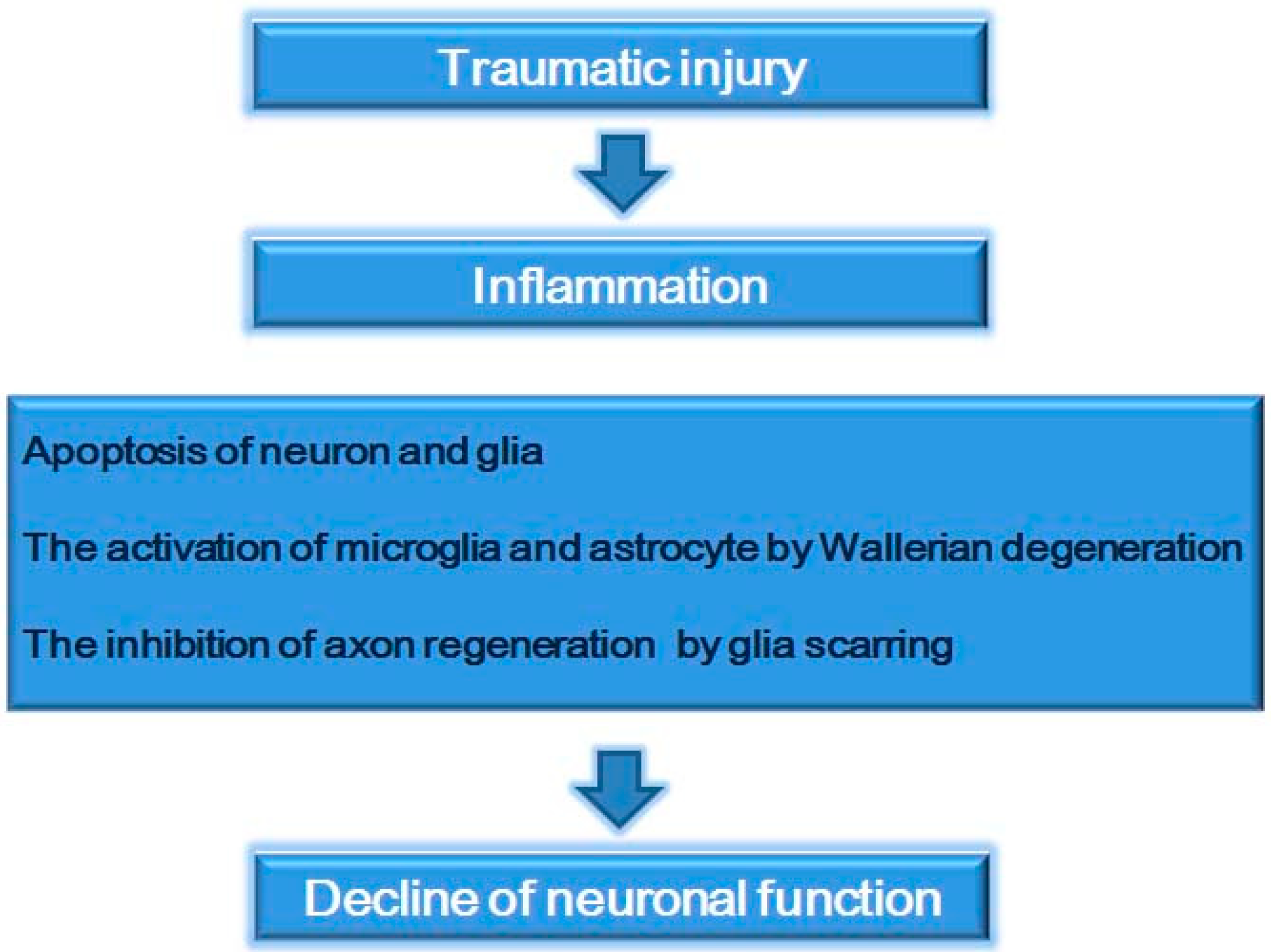
2. Spinal Cord Injury

3. RAGE
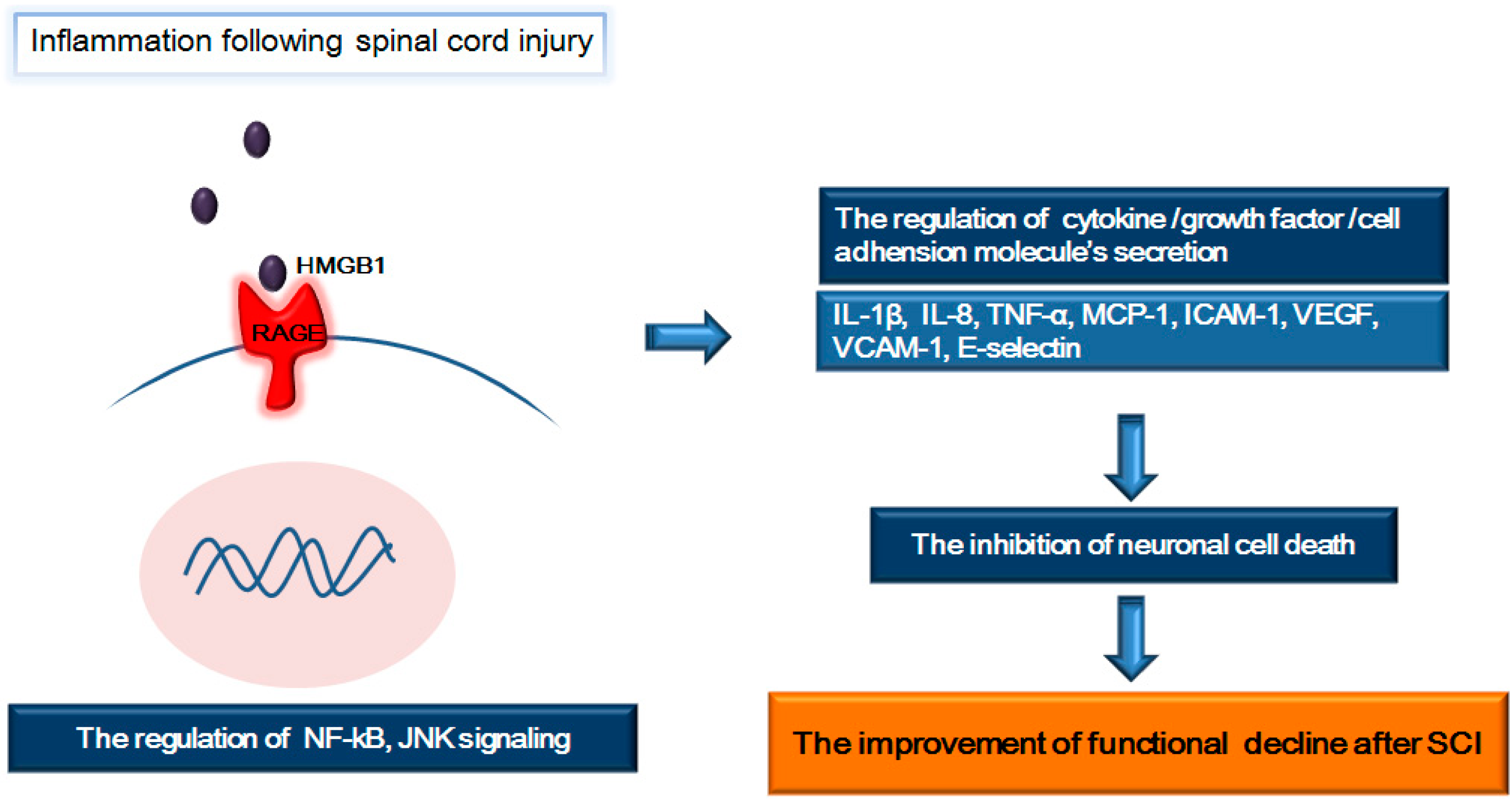
4. RAGE and Its Ligands (HMGB1 and S100β): Focus on Inflammation Following SCI

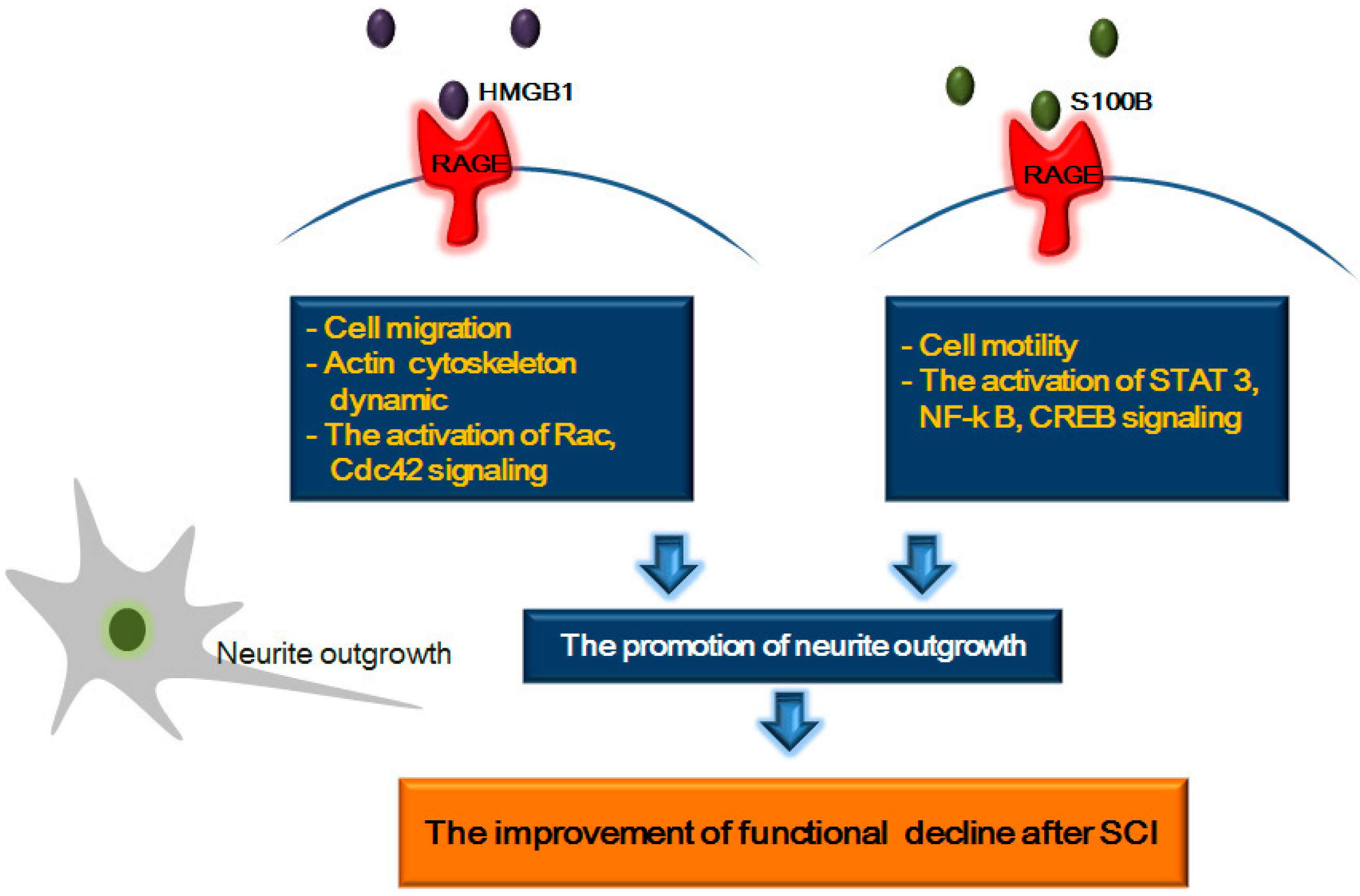
5. RAGE and Its Ligands: Focus on Neurite Outgrowth Following SCI

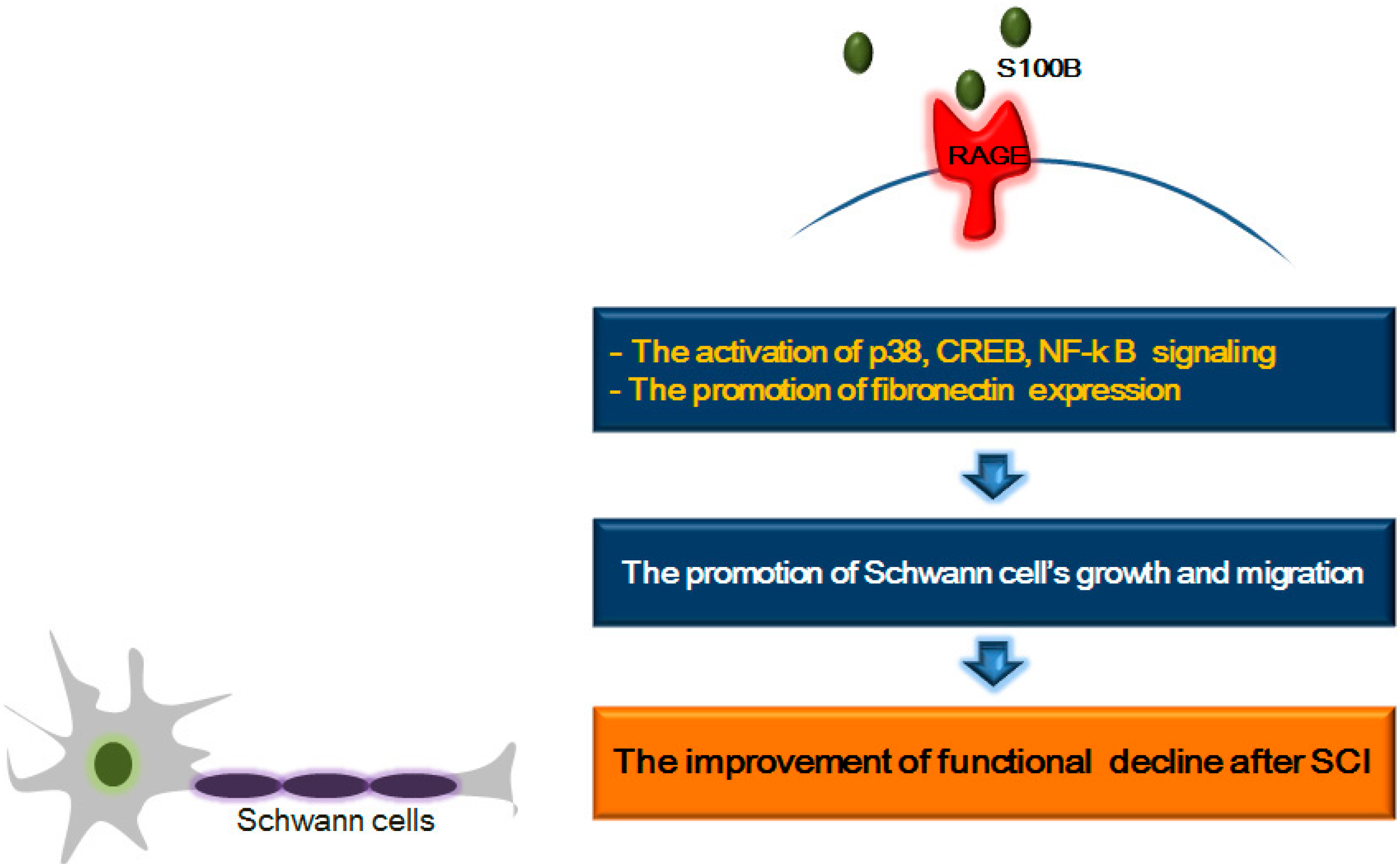
6. RAGE and Its Ligands: Focus on Schwann Cell Growth and Migration after SCI

7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scott, J.M.; Warburton, D.E.; Williams, D.; Whelan, S.; Krassioukov, A. Challenges, concerns and common problems: Physiological consequences of spinal cord injury and microgravity. Spinal Cord 2011, 49, 4–16. [Google Scholar] [CrossRef]
- Thuret, S.; Moon, L.D.; Gage, F.H. Therapeutic interventions after spinal cord injury. Nat. Rev. Neurosci. 2006, 7, 628–643. [Google Scholar] [CrossRef]
- Teng, Y.D.; Lavik, E.B.; Qu, X.; Park, K.I.; Ourednik, J.; Zurakowski, D.; Langer, R.; Snyder, E.Y. Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 3024–3029. [Google Scholar]
- Lopez-Gonzalez, R.; Velasco, I. Therapeutic potential of motor neurons differentiated from embryonic stem cells and induced pluripotent stem cells. Arch. Med. Res. 2012, 43, 1–10. [Google Scholar]
- Pearse, D.D.; Marcillo, A.E.; Oudega, M.; Lynch, M.P.; Wood, P.M.; Bunge, M.B. Transplantation of Schwann cells and olfactory ensheathing glia after spinal cord injury: Does pretreatment with methylprednisolone and interleukin-10 enhance recovery? J. Neurotrauma 2004, 21, 1223–1239. [Google Scholar] [CrossRef]
- Oudega, M.; Xu, X.M. Schwann cell transplantation for repair of the adult spinal cord. J. Neurotrauma 2006, 23, 453–467. [Google Scholar] [CrossRef]
- Hendriks, W.T.; Ruitenberg, M.J.; Blits, B.; Boer, G.J.; Verhaagen, J. Viral vector-mediated gene transfer of neurotrophins to promote regeneration of the injured spinal cord. Prog. Brain Res. 2004, 146, 451–476. [Google Scholar] [CrossRef]
- Taylor, S.J.; McDonald, J.W., 3rd; Sakiyama-Elbert, S.E. Controlled release of neurotrophin-3 from fibrin gels for spinal cord injury. J. Control. Release 2004, 98, 281–294. [Google Scholar]
- Bethea, J.R.; Nagashima, H.; Acosta, M.C.; Briceno, C.; Gomez, F.; Marcillo, A.E.; Loor, K.; Green, J.; Dietrich, W.D. Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J. Neurotrauma 1999, 16, 851–863. [Google Scholar] [CrossRef]
- Gok, B.; Sciubba, D.M.; Okutan, O.; Beskonakli, E.; Palaoglu, S.; Erdamar, H.; Sargon, M.F. Immunomodulation of acute experimental spinal cord injury with human immunoglobulin G. J. Clin. Neurosci. 2009, 16, 549–553. [Google Scholar] [CrossRef]
- Fleming, J.C.; Norenberg, M.D.; Ramsay, D.A.; Dekaban, G.A.; Marcillo, A.E.; Saenz, A.D.; Pasquale-Styles, M.; Dietrich, W.D.; Weaver, L.C. The cellular inflammatory response in human spinal cords after injury. Brain 2006, 129, 3249–3269. [Google Scholar] [CrossRef]
- Nakahara, S.; Yone, K.; Sakou, T.; Wada, S.; Nagamine, T.; Niiyama, T.; Ichijo, H. Induction of apoptosis signal regulating kinase 1 (ASK1) after spinal cord injury in rats: Possible involvement of ASK1-JNK and -p38 pathways in neuronal apoptosis. J. Neuropathol. Exp. Neurol. 1999, 58, 442–450. [Google Scholar] [CrossRef]
- Liu, S.; Ruenes, G.L.; Yezierski, R.P. NMDA and non-NMDA receptor antagonists protect against excitotoxic injury in the rat spinal cord. Brain Res. 1997, 756, 160–167. [Google Scholar] [CrossRef]
- Kato, H.; Kanellopoulos, G.K.; Matsuo, S.; Wu, Y.J.; Jacquin, M.F.; Hsu, C.Y.; Kouchoukos, N.T.; Choi, D.W. Neuronal apoptosis and necrosis following spinal cord ischemia in the rat. Exp. Neurol. 1997, 148, 464–474. [Google Scholar] [CrossRef]
- Crowe, M.J.; Bresnahan, J.C.; Shuman, S.L.; Masters, J.N.; Beattie, M.S. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat. Med. 1997, 3, 73–76. [Google Scholar] [CrossRef]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef]
- Mizuno, Y.; Mochizuki, H.; Sugita, Y.; Goto, K. Apoptosis in neurodegenerative disorders. Intern. Med. 1998, 37, 192–193. [Google Scholar] [CrossRef]
- Byrnes, K.R.; Stoica, B.A.; Fricke, S.; di Giovanni, S.; Faden, A.I. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain 2007, 130, 2977–2992. [Google Scholar] [CrossRef]
- Stern, D.; Yan, S.D.; Yan, S.F.; Schmidt, A.M. Receptor for advanced glycation endproducts: A multiligand receptor magnifying cell stress in diverse pathologic settings. Adv. Drug Deliv. Rev. 2002, 54, 1615–1625. [Google Scholar] [CrossRef]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef]
- Donato, R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Donato, R. RAGE: A single receptor for several ligands and different cellular responses: The case of certain S100 proteins. Curr. Mol. Med. 2007, 7, 711–724. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The RAGE axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ. Res. 2010, 106, 842–853. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- D’Agati, V.; Schmidt, A.M. RAGE and the pathogenesis of chronic kidney disease. Nat. Rev. Nephrol. 2010, 6, 352–360. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Kamide, T.; Kitao, Y.; Takeichi, T.; Okada, A.; Mohri, H.; Schmidt, A.M.; Kawano, T.; Munesue, S.; Yamamoto, Y.; Yamamoto, H.; et al. RAGE mediates vascular injury and inflammation after global cerebral ischemia. Neurochem. Int. 2012, 60, 220–228. [Google Scholar] [CrossRef]
- Jin, Q.; Chen, H.; Luo, A.; Ding, F.; Liu, Z. S100A14 stimulates cell proliferation and induces cell apoptosis at different concentrations via receptor for advanced glycation end products (RAGE). PLoS One 2011, 6, e19375. [Google Scholar]
- Tator, C.H.; Fehlings, M.G. Review of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J. Neurosurg. 1991, 75, 15–26. [Google Scholar] [CrossRef]
- Dumont, R.J.; Verma, S.; Okonkwo, D.O.; Hurlbert, R.J.; Boulos, P.T.; Ellegala, D.B.; Dumont, A.S. Acute spinal cord injury, part II: Contemporary pharmacotherapy. Clin. Neuropharmacol. 2001, 24, 265–279. [Google Scholar] [CrossRef]
- Whalley, K.; O’Neill, P.; Ferretti, P. Changes in response to spinal cord injury with development: Vascularization, hemorrhage and apoptosis. Neuroscience 2006, 137, 821–832. [Google Scholar] [CrossRef]
- McDonald, J.W.; Sadowsky, C. Spinal-cord injury. Lancet 2002, 359, 417–425. [Google Scholar] [CrossRef]
- Rosenfeld, J.V.; Bandopadhayay, P.; Goldschlager, T.; Brown, D.J. The ethics of the treatment of spinal cord injury: Stem cell transplants, motor neuroprosthetics, and social equity. Top. Spinal Cord Inj. Rehabil. 2008, 14, 76–88. [Google Scholar]
- Genovese, T.; Esposito, E.; Mazzon, E.; di Paola, R.; Caminiti, R.; Bramanti, P.; Cappelani, A.; Cuzzocrea, S. Absence of endogenous interleukin-10 enhances secondary inflammatory process after spinal cord compression injury in mice. J. Neurochem. 2009, 108, 1360–1372. [Google Scholar] [CrossRef]
- Shen, L.F.; Cheng, H.; Tsai, M.C.; Kuo, H.S.; Chak, K.F. PAL31 may play an important role as inflammatory modulator in the repair process of the spinal cord injury rat. J. Neurochem. 2009, 108, 1187–1197. [Google Scholar] [CrossRef]
- Pineau, I.; Lacroix, S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: Multiphasic expression pattern and identification of the cell types involved. J. Comp. Neurol. 2007, 500, 267–285. [Google Scholar] [CrossRef]
- Yang, L.; Jones, N.R.; Blumbergs, P.C.; van den Heuvel, C.; Moore, E.J.; Manavis, J.; Sarvestani, G.T.; Ghabriel, M.N. Severity-dependent expression of pro-inflammatory cytokines in traumatic spinal cord injury in the rat. J. Clin. Neurosci. 2005, 12, 276–284. [Google Scholar] [CrossRef]
- Yang, L.; Blumbergs, P.C.; Jones, N.R.; Manavis, J.; Sarvestani, G.T.; Ghabriel, M.N. Early expression and cellular localization of proinflammatory cytokines interleukin-1β, interleukin-6, and tumor necrosis factor-α in human traumatic spinal cord injury. Spine 2004, 29, 966–971. [Google Scholar] [CrossRef]
- Schnell, L.; Fearn, S.; Schwab, M.E.; Perry, V.H.; Anthony, D.C. Cytokine-induced acute inflammation in the brain and spinal cord. J. Neuropathol. Exp. Neurol. 1999, 58, 245–254. [Google Scholar] [CrossRef]
- Cai, Z.H.; Song, L.S.; Gao, C.P.; Wu, L.T.; Qiu, L.H.; Xiang, J.H. Cloning and expression of tumor necrosis factor (TNFα) cDNA from red seabream pagrus major. ShengWu HuaXue Yu ShengWu WuLi XueBao 2003, 35, 1111–1116. [Google Scholar]
- Hermann, G.E.; Rogers, R.C.; Bresnahan, J.C.; Beattie, M.S. Tumor necrosis factor-α induces cFOS and strongly potentiates glutamate-mediated cell death in the rat spinal cord. Neurobiol. Dis. 2001, 8, 590–599. [Google Scholar] [CrossRef]
- Lee, Y.B.; Yune, T.Y.; Baik, S.Y.; Shin, Y.H.; Du, S.; Rhim, H.; Lee, E.B.; Kim, Y.C.; Shin, M.L.; Markelonis, G.J.; et al. Role of tumor necrosis factor-alpha in neuronal and glial apoptosis after spinal cord injury. Exp. Neurol. 2000, 166, 190–195. [Google Scholar]
- Lu, K.T.; Wang, Y.W.; Yang, J.T.; Yang, Y.L.; Chen, H.I. Effect of interleukin-1 on traumatic brain injury-induced damage to hippocampal neurons. J. Neurotrauma 2005, 22, 885–895. [Google Scholar] [CrossRef]
- Shamash, S.; Reichert, F.; Rotshenker, S. The cytokine network of Wallerian degeneration: Tumor necrosis factor-α, interleukin-1α, and interleukin-1β. J. Neurosci. 2002, 22, 3052–3060. [Google Scholar]
- Streit, W.J.; Semple-Rowland, S.L.; Hurley, S.D.; Miller, R.C.; Popovich, P.G.; Stokes, B.T. Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp. Neurol. 1998, 152, 74–87. [Google Scholar]
- Dusart, I.; Schwab, M.E. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur. J. Neurosci. 1994, 6, 712–724. [Google Scholar] [CrossRef]
- Fujiki, M.; Zhang, Z.; Guth, L.; Steward, O. Genetic influences on cellular reactions to spinal cord injury: Activation of macrophages/microglia and astrocytes is delayed in mice carrying a mutation (WldS) that causes delayed Wallerian degeneration. J. Comp. Neurol. 1996, 371, 469–484. [Google Scholar]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Schwab, M.E. Repairing the injured spinal cord. Science 2002, 295, 1029–1031. [Google Scholar] [CrossRef]
- Yuan, Y.M.; He, C. The glial scar in spinal cord injury and repair. Neurosci. Bull. 2013, 29, 421–435. [Google Scholar] [CrossRef]
- Gensel, J.C.; Nakamura, S.; Guan, Z.; van Rooijen, N.; Ankeny, D.P.; Popovich, P.G. Macrophages promote axon regeneration with concurrent neurotoxicity. J. Neurosci. 2009, 29, 3956–3968. [Google Scholar] [CrossRef]
- Anthes, D.L.; Theriault, E.; Tator, C.H. Characterization of axonal ultrastructural pathology following experimental spinal cord compression injury. Brain Res. 1995, 702, 1–16. [Google Scholar] [CrossRef]
- Muradov, J.M.; Hagg, T. Intravenous infusion of magnesium chloride improves epicenter blood flow during the acute stage of contusive spinal cord injury in rats. J. Neurotrauma 2013, 30, 840–852. [Google Scholar] [CrossRef]
- Tator, C.H.; Koyanagi, I. Vascular mechanisms in the pathophysiology of human spinal cord injury. J. Neurosurg. 1997, 86, 483–492. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Stern, D.M. RAGE: A new target for the prevention and treatment of the vascular and inflammatory complications of diabetes. Trends Endocrinol. Metab. 2000, 11, 368–375. [Google Scholar] [CrossRef]
- Freigang, J.; Proba, K.; Leder, L.; Diederichs, K.; Sonderegger, P.; Welte, W. The crystal structure of the ligand binding module of axonin-1/TAG-1 suggests a zipper mechanism for neural cell adhesion. Cell 2000, 101, 425–433. [Google Scholar] [CrossRef]
- Soroka, V.; Kolkova, K.; Kastrup, J.S.; Diederichs, K.; Breed, J.; Kiselyov, V.V.; Poulsen, F.M.; Larsen, I.K.; Welte, W.; Berezin, V.; et al. Structure and interactions of NCAM Ig1-2-3 suggest a novel zipper mechanism for homophilic adhesion. Structure 2003, 11, 1291–1301. [Google Scholar] [CrossRef]
- Ramasamy, R.; Vannucci, S.J.; Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar]
- Yamamoto, Y.; Kato, I.; Doi, T.; Yonekura, H.; Ohashi, S.; Takeuchi, M.; Watanabe, T.; Yamagishi, S.; Sakurai, S.; Takasawa, S.; et al. Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J. Clin. Investig. 2001, 108, 261–268. [Google Scholar] [CrossRef]
- Penfold, S.A.; Coughlan, M.T.; Patel, S.K.; Srivastava, P.M.; Sourris, K.C.; Steer, D.; Webster, D.E.; Thomas, M.C.; MacIsaac, R.J.; Jerums, G.; et al. Circulating high-molecular-weight RAGE ligands activate pathways implicated in the development of diabetic nephropathy. Kidney Int. 2010, 78, 287–295. [Google Scholar] [CrossRef]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Nemeth, J.; Furstenberger, G.; Muller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef]
- Riehl, A.; Nemeth, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef]
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Fages, C.; Nolo, R.; Huttunen, H.J.; Eskelinen, E.; Rauvala, H. Regulation of cell migration by amphoterin. J. Cell Sci. 2000, 113, 611–620. [Google Scholar]
- Van Beijnum, J.R.; Buurman, W.A.; Griffioen, A.W. Convergence and amplification of toll-like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 2008, 11, 91–99. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Schafer, B.W.; Sasaki, N.; Heizmann, C.W. Expression analysis of S100 proteins and RAGE in human tumors using tissue microarrays. Biochem. Biophys. Res. Commun. 2003, 307, 375–381. [Google Scholar]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Zlokovic, B.V. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics 2008, 5, 409–414. [Google Scholar] [CrossRef]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Soluble RAGE: Therapy and biomarker in unraveling the RAGE axis in chronic disease and aging. Biochem. Pharmacol. 2010, 79, 1379–1386. [Google Scholar] [CrossRef]
- Candela, P.; Gosselet, F.; Saint-Pol, J.; Sevin, E.; Boucau, M.C.; Boulanger, E.; Cecchelli, R.; Fenart, L. Apical-to-basolateral transport of amyloid-β peptides through blood-brain barrier cells is mediated by the receptor for advanced glycation end-products and is restricted by P-glycoprotein. J. Alzheimer’s Dis. 2010, 22, 849–859. [Google Scholar]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta 2009, 1793, 993–1007. [Google Scholar] [CrossRef]
- Yan, S.S.; Wu, Z.Y.; Zhang, H.P.; Furtado, G.; Chen, X.; Yan, S.F.; Schmidt, A.M.; Brown, C.; Stern, A.; LaFaille, J.; et al. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat. Med. 2003, 9, 287–293. [Google Scholar] [CrossRef]
- Matsumoto, S.; Yoshida, T.; Murata, H.; Harada, S.; Fujita, N.; Nakamura, S.; Yamamoto, Y.; Watanabe, T.; Yonekura, H.; Yamamoto, H.; et al. Solution structure of the variable-type domain of the receptor for advanced glycation end products: New insight into AGE-RAGE interaction. Biochemistry 2008, 47, 12299–12311. [Google Scholar] [CrossRef]
- Koch, M.; Chitayat, S.; Dattilo, B.M.; Schiefner, A.; Diez, J.; Chazin, W.J.; Fritz, G. Structural basis for ligand recognition and activation of RAGE. Structure 2010, 18, 1342–1352. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Manfredi, A.A. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol. Rev. 2007, 220, 35–46. [Google Scholar] [CrossRef]
- Rauvala, H.; Rouhiainen, A. RAGE as a receptor of HMGB1 (Amphoterin): Roles in health and disease. Curr. Mol. Med. 2007, 7, 725–734. [Google Scholar] [CrossRef]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar]
- Sakaguchi, T.; Yan, S.F.; Yan, S.D.; Belov, D.; Rong, L.L.; Sousa, M.; Andrassy, M.; Marso, S.P.; Duda, S.; Arnold, B.; et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J. Clin. Investig. 2003, 111, 959–972. [Google Scholar] [CrossRef]
- Han, S.H.; Kim, Y.H.; Mook-Jung, I. RAGE: The beneficial and deleterious effects by diverse mechanisms of actions. Mol. Cells 2011, 31, 91–97. [Google Scholar] [CrossRef]
- Hudson, B.I.; Kalea, A.Z.; del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. RAGE: Therapeutic target and biomarker of the inflammatory response—The evidence mounts. J. Leukoc. Biol. 2009, 86, 505–512. [Google Scholar] [CrossRef]
- Xu, Y.; Toure, F.; Qu, W.; Lin, L.; Song, F.; Shen, X.; Rosario, R.; Garcia, J.; Schmidt, A.M.; Yan, S.F. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J. Biol. Chem. 2010, 285, 23233–23240. [Google Scholar] [CrossRef]
- Bierhaus, A.; Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Kloting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-κB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef]
- Bierhaus, A.; Stern, D.M.; Nawroth, P.P. RAGE in inflammation: A new therapeutic target? Curr. Opin. Investig. Drugs 2006, 7, 985–991. [Google Scholar]
- Chen, K.B.; Uchida, K.; Nakajima, H.; Yayama, T.; Hirai, T.; Rodriguez Guerrero, A.; Kobayashi, S.; Ma, W.Y.; Liu, S.Y.; Zhu, P.; et al. High-mobility group box-1 and its receptors contribute to proinflammatory response in the acute phase of spinal cord injury in rats. Spine 2011, 36, 2122–2129. [Google Scholar] [CrossRef]
- Kawabata, H.; Setoguchi, T.; Yone, K.; Souda, M.; Yoshida, H.; Kawahara, K.; Maruyama, I.; Komiya, S. High mobility group box 1 is upregulated after spinal cord injury and is associated with neuronal cell apoptosis. Spine 2010, 35, 1109–1115. [Google Scholar]
- Guo, J.D.; Li, L.; Shi, Y.M.; Wang, H.D.; Yuan, Y.L.; Shi, X.X.; Hou, S.X. Genetic ablation of receptor for advanced glycation end products promotes functional recovery in mouse model of spinal cord injury. Mol. Cell. Biochem. 2014, 390, 215–223. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, M.J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar]
- Xu, D.; Young, J.; Song, D.; Esko, J.D. Heparan sulfate is essential for high mobility group protein 1 (HMGB1) signaling by the receptor for advanced glycation end products (RAGE). J. Biol. Chem. 2011, 286, 41736–41744. [Google Scholar] [CrossRef]
- Kislinger, T.; Fu, C.; Huber, B.; Qu, W.; Taguchi, A.; Du Yan, S.; Hofmann, M.; Yan, S.F.; Pischetsrieder, M.; Stern, D.; et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression . J. Biol. Chem. 1999, 274, 31740–31749. [Google Scholar] [CrossRef]
- Guazzi, S.; Strangio, A.; Franzi, A.T.; Bianchi, M.E. HMGB1, an architectural chromatin protein and extracellular signalling factor, has a spatially and temporally restricted expression pattern in mouse brain. Gene Expr. Patterns 2003, 3, 29–33. [Google Scholar] [CrossRef]
- Enokido, Y.; Yoshitake, A.; Ito, H.; Okazawa, H. Age-dependent change of HMGB1 and DNA double-strand break accumulation in mouse brain. Biochem. Biophys. Res. Commun. 2008, 376, 128–133. [Google Scholar]
- Daston, M.M.; Ratner, N. Amphoterin (P30, HMG-1) and RIP are early markers of oligodendrocytes in the developing rat spinal cord. J. Neurocytol. 1994, 23, 323–332. [Google Scholar] [CrossRef]
- Tenenbaum, T.; Essmann, F.; Adam, R.; Seibt, A.; Janicke, R.U.; Novotny, G.E.; Galla, H.J.; Schroten, H. Cell death, caspase activation, and HMGB1 release of porcine choroid plexus epithelial cells during Streptococcus suis infection in vitro. Brain Res. 2006, 1100, 1–12. [Google Scholar] [CrossRef]
- Gao, H.M.; Zhou, H.; Zhang, F.; Wilson, B.C.; Kam, W.; Hong, J.S. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J. Neurosci. 2011, 31, 1081–1092. [Google Scholar] [CrossRef]
- Daston, M.M.; Ratner, N. Expression of P30, a protein with adhesive properties, in Schwann cells and neurons of the developing and regenerating peripheral nerve. J. Cell Biol. 1991, 112, 1229–1239. [Google Scholar] [CrossRef]
- Adami, C.; Sorci, G.; Blasi, E.; Agneletti, A.L.; Bistoni, F.; Donato, R. S100B expression in and effects on microglia. Glia 2001, 33, 131–142. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Mechanisms of disease: Advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 285–293. [Google Scholar] [CrossRef]
- Meneghini, V.; Francese, M.T.; Carraro, L.; Grilli, M. A novel role for the Receptor for Advanced Glycation End-products in neural progenitor cells derived from adult SubVentricular Zone. Mol. Cell. Neurosci. 2010, 45, 139–150. [Google Scholar] [CrossRef]
- Liu, X.Z.; Xu, X.M.; Hu, R.; Du, C.; Zhang, S.X.; McDonald, J.W.; Dong, H.X.; Wu, Y.J.; Fan, G.S.; Jacquin, M.F.; et al. Neuronal and glial apoptosis after traumatic spinal cord injury. J. Neurosci. 1997, 17, 5395–5406. [Google Scholar]
- Kwon, B.K.; Oxland, T.R.; Tetzlaff, W. Animal models used in spinal cord regeneration research. Spine 2002, 27, 1504–1510. [Google Scholar] [CrossRef]
- Kwon, B.K.; Tetzlaff, W.; Grauer, J.N.; Beiner, J.; Vaccaro, A.R. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004, 4, 451–464. [Google Scholar] [CrossRef]
- Norenberg, M.D.; Smith, J.; Marcillo, A. The pathology of human spinal cord injury: Defining the problems. J. Neurotrauma 2004, 21, 429–440. [Google Scholar] [CrossRef]
- Foote, A.K.; Blakemore, W.F. Inflammation stimulates remyelination in areas of chronic demyelination. Brain 2005, 128, 528–539. [Google Scholar] [CrossRef]
- Ludwin, S.K. Chronic demyelination inhibits remyelination in the central nervous system. An analysis of contributing factors. Lab. Investig. 1980, 43, 382–387. [Google Scholar]
- Miron, V.E.; Boyd, A.; Zhao, J.W.; Yuen, T.J.; Ruckh, J.M.; Shadrach, J.L.; van Wijngaarden, P.; Wagers, A.J.; Williams, A.; Franklin, R.J.; et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013, 16, 1211–1218. [Google Scholar] [CrossRef]
- Morell, P.; Barrett, C.V.; Mason, J.L.; Toews, A.D.; Hostettler, J.D.; Knapp, G.W.; Matsushima, G.K. Gene expression in brain during cuprizone-induced demyelination and remyelination. Mol. Cell. Neurosci. 1998, 12, 220–227. [Google Scholar] [CrossRef]
- Katoh, S.; Ikata, T.; Tsubo, M.; Hamada, Y.; el Masry, W.S. Possible implication of leukocytes in secondary pathological changes after spinal cord injury. Injury 1997, 28, 215–217. [Google Scholar] [CrossRef]
- Li, Z.; Hogan, E.L.; Banik, N.L. Role of calpain in spinal cord injury: Increased calpain immunoreactivity in rat spinal cord after impact trauma. Neurochem. Res. 1996, 21, 441–448. [Google Scholar] [CrossRef]
- Lu, J.; Ashwell, K.W.; Waite, P. Advances in secondary spinal cord injury: Role of apoptosis. Spine 2000, 25, 1859–1866. [Google Scholar] [CrossRef]
- Habgood, M.D.; Bye, N.; Dziegielewska, K.M.; Ek, C.J.; Lane, M.A.; Potter, A.; Morganti-Kossmann, C.; Saunders, N.R. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur. J. Neurosci. 2007, 25, 231–238. [Google Scholar] [CrossRef]
- Han, X.; Lu, M.; Wang, S.; Lv, D.; Liu, H. Targeting IKK/NF-κB pathway reduces infiltration of inflammatory cells and apoptosis after spinal cord injury in rats. Neurosci. Lett. 2012, 511, 28–32. [Google Scholar] [CrossRef]
- Brambilla, R.; Bracchi-Ricard, V.; Hu, W.H.; Frydel, B.; Bramwell, A.; Karmally, S.; Green, E.J.; Bethea, J.R. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J. Exp. Med. 2005, 202, 145–156. [Google Scholar] [CrossRef]
- Brambilla, R.; Persaud, T.; Hu, X.; Karmally, S.; Shestopalov, V.I.; Dvoriantchikova, G.; Ivanov, D.; Nathanson, L.; Barnum, S.R.; Bethea, J.R. Transgenic inhibition of astroglial NF-κB improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J. Immunol. 2009, 182, 2628–2640. [Google Scholar]
- Yaser, A.M.; Huang, Y.; Zhou, R.R.; Hu, G.S.; Xiao, M.F.; Huang, Z.B.; Duan, C.J.; Tian, W.; Tang, D.L.; Fan, X.G. The role of receptor for advanced glycation end products (RAGE) in the proliferation of hepatocellular carcinoma. Int. J. Mol. Sci. 2012, 13, 5982–5997. [Google Scholar] [CrossRef]
- Huang, Y.; Xie, K.; Li, J.; Xu, N.; Gong, G.; Wang, G.; Yu, Y.; Dong, H.; Xiong, L. Beneficial effects of hydrogen gas against spinal cord ischemia-reperfusion injury in rabbits. Brain Res. 2011, 1378, 125–136. [Google Scholar] [CrossRef]
- Wang, Q.; Ding, Q.; Zhou, Y.; Gou, X.; Hou, L.; Chen, S.; Zhu, Z.; Xiong, L. Ethyl pyruvate attenuates spinal cord ischemic injury with a wide therapeutic window through inhibiting high-mobility group box 1 release in rabbits. Anesthesiology 2009, 110, 1279–1286. [Google Scholar] [CrossRef]
- Esposito, E.; Genovese, T.; Caminiti, R.; Bramanti, P.; Meli, R.; Cuzzocrea, S. Melatonin reduces stress-activated/mitogen-activated protein kinases in spinal cord injury. J. Pineal Res. 2009, 46, 79–86. [Google Scholar] [CrossRef]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde, A.; Yan, S.F.; Stern, A.; et al. RAGE potentiates Aβ-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004, 23, 4096–4105. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar]
- Vincent, A.M.; Perrone, L.; Sullivan, K.A.; Backus, C.; Sastry, A.M.; Lastoskie, C.; Feldman, E.L. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology 2007, 148, 548–558. [Google Scholar] [CrossRef]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Pisetsky, D.S.; Erlandsson-Harris, H.; Andersson, U. High-mobility group box protein 1 (HMGB1): An alarmin mediating the pathogenesis of rheumatic disease. Arthritis Res. Ther. 2008, 10, 209. [Google Scholar] [CrossRef]
- Dumitriu, I.E.; Baruah, P.; Valentinis, B.; Voll, R.E.; Herrmann, M.; Nawroth, P.P.; Arnold, B.; Bianchi, M.E.; Manfredi, A.A.; Rovere-Querini, P. Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 2005, 174, 7506–7515. [Google Scholar] [CrossRef]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef]
- Andersson, A.; Covacu, R.; Sunnemark, D.; Danilov, A.I.; dal Bianco, A.; Khademi, M.; Wallstrom, E.; Lobell, A.; Brundin, L.; Lassmann, H.; et al. Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J. Leukoc. Biol. 2008, 84, 1248–1255. [Google Scholar] [CrossRef]
- Lin, L. RAGE on the Toll Road? Cell Mol. Immunol. 2006, 3, 351–358. [Google Scholar]
- Ruan, B.H.; Li, X.; Winkler, A.R.; Cunningham, K.M.; Kuai, J.; Greco, R.M.; Nocka, K.H.; Fitz, L.J.; Wright, J.F.; Pittman, D.D.; et al. Complement C3a, CpG oligos, and DNA/C3a complex stimulate IFN-α production in a receptor for advanced glycation end product-dependent manner. J. Immunol. 2010, 185, 4213–4222. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Harashima, A.; Saito, H.; Tsuneyama, K.; Munesue, S.; Motoyoshi, S.; Han, D.; Watanabe, T.; Asano, M.; Takasawa, S.; et al. Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J. Immunol. 2011, 186, 3248–3257. [Google Scholar] [CrossRef]
- Kawabata, D.; Venkatesh, J.; Ramanujam, M.; Davidson, A.; Grimaldi, C.M.; Diamond, B. Enhanced selection of high affinity DNA-reactive B cells following cyclophosphamide treatment in mice. PLoS One 2010, 5, e8418. [Google Scholar]
- Chen, G.; Ward, M.F.; Sama, A.E.; Wang, H. Extracellular HMGB1 as a proinflammatory cytokine. J. Interferon Cytokine Res. 2004, 24, 329–333. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Tracey, K.J. HMG-1 rediscovered as a cytokine. Shock 2001, 15, 247–253. [Google Scholar] [CrossRef]
- Wang, H.; Yang, H.; Czura, C.J.; Sama, A.E.; Tracey, K.J. HMGB1 as a late mediator of lethal systemic inflammation. Am. J. Respir. Crit. Care Med. 2001, 164, 1768–1773. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Czura, C.J.; Tracey, K.J. HMGB1 as a cytokine and therapeutic target. J. Endotoxin Res. 2002, 8, 469–472. [Google Scholar] [CrossRef]
- Dumitriu, I.E.; Bianchi, M.E.; Bacci, M.; Manfredi, A.A.; Rovere-Querini, P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J. Leukoc. Biol. 2007, 81, 84–91. [Google Scholar]
- Yang, D.; Chen, Q.; Yang, H.; Tracey, K.J.; Bustin, M.; Oppenheim, J.J. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J. Leukoc. Biol. 2007, 81, 59–66. [Google Scholar]
- Messmer, D.; Yang, H.; Telusma, G.; Knoll, F.; Li, J.; Messmer, B.; Tracey, K.J.; Chiorazzi, N. High mobility group box protein 1: An endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 2004, 173, 307–313. [Google Scholar]
- Rovere-Querini, P.; Capobianco, A.; Scaffidi, P.; Valentinis, B.; Catalanotti, F.; Giazzon, M.; Dumitriu, I.E.; Muller, S.; Iannacone, M.; Traversari, C.; et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004, 5, 825–830. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- Park, J.S.; Gamboni-Robertson, F.; He, Q.; Svetkauskaite, D.; Kim, J.Y.; Strassheim, D.; Sohn, J.W.; Yamada, S.; Maruyama, I.; Banerjee, A.; et al. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell Physiol. 2006, 290, C917–C924. [Google Scholar]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar]
- Park, J.S.; Arcaroli, J.; Yum, H.K.; Yang, H.; Wang, H.; Yang, K.Y.; Choe, K.H.; Strassheim, D.; Pitts, T.M.; Tracey, K.J.; et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am. J. Physiol. Cell Physiol. 2003, 284, C870–C879. [Google Scholar] [CrossRef]
- Vajn, K.; Plunkett, J.A.; Tapanes-Castillo, A.; Oudega, M. Axonal regeneration after spinal cord injury in zebrafish and mammals: Differences, similarities, translation. Neurosci. Bull. 2013, 29, 402–410. [Google Scholar] [CrossRef]
- Cao, Q.; Xu, X.M.; Devries, W.H.; Enzmann, G.U.; Ping, P.; Tsoulfas, P.; Wood, P.M.; Bunge, M.B.; Whittemore, S.R. Functional recovery in traumatic spinal cord injury after transplantation of multineurotrophin-expressing glial-restricted precursor cells. J. Neurosci. 2005, 25, 6947–6957. [Google Scholar] [CrossRef]
- Cummings, B.J.; Uchida, N.; Tamaki, S.J.; Salazar, D.L.; Hooshmand, M.; Summers, R.; Gage, F.H.; Anderson, A.J. Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice. Proc. Natl. Acad. Sci. USA 2005, 102, 14069–14074. [Google Scholar] [CrossRef]
- Hofstetter, C.P.; Holmstrom, N.A.; Lilja, J.A.; Schweinhardt, P.; Hao, J.; Spenger, C.; Wiesenfeld-Hallin, Z.; Kurpad, S.N.; Frisen, J.; Olson, L. Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nature Neurosci. 2005, 8, 346–353. [Google Scholar] [CrossRef]
- Karimi-Abdolrezaee, S.; Eftekharpour, E.; Wang, J.; Morshead, C.M.; Fehlings, M.G. Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. J. Neurosci. 2006, 26, 3377–3389. [Google Scholar] [CrossRef]
- Lee, K.H.; Yoon, D.H.; Park, Y.G.; Lee, B.H. Effects of glial transplantation on functional recovery following acute spinal cord injury. J. Neurotrauma 2005, 22, 575–589. [Google Scholar] [CrossRef]
- Mitsui, T.; Shumsky, J.S.; Lepore, A.C.; Murray, M.; Fischer, I. Transplantation of neuronal and glial restricted precursors into contused spinal cord improves bladder and motor functions, decreases thermal hypersensitivity, and modifies intraspinal circuitry. J. Neurosci. 2005, 25, 9624–9636. [Google Scholar] [CrossRef]
- Rauvala, H.; Pihlaskari, R. Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J. Biol. Chem. 1987, 262, 16625–16635. [Google Scholar]
- Zhao, Z.; Nair, S.M.; Chou, D.K.; Tobet, S.A.; Jungalwala, F.B. Expression and role of sulfoglucuronyl (HNK-1) carbohydrate and its binding protein SBP-1 in developing rat cerebral cortex. J. Neurosci. Res. 2000, 62, 186–205. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Rauvala, H. Amphoterin as an extracellular regulator of cell motility: From discovery to disease. J. Intern. Med. 2004, 255, 351–366. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Rauvala, H. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J. Biol. Chem. 2002, 277, 38635–38646. [Google Scholar]
- Srikrishna, G.; Huttunen, H.J.; Johansson, L.; Weigle, B.; Yamaguchi, Y.; Rauvala, H.; Freeze, H.H. N-Glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J. Neurochem. 2002, 80, 998–1008. [Google Scholar] [CrossRef]
- Rauvala, H.; Huttunen, H.J.; Fages, C.; Kaksonen, M.; Kinnunen, T.; Imai, S.; Raulo, E.; Kilpelainen, I. Heparin-binding proteins HB-GAM (pleiotrophin) and amphoterin in the regulation of cell motility. Matrix Biol. 2000, 19, 377–387. [Google Scholar] [CrossRef]
- Reddy, M.A.; Li, S.L.; Sahar, S.; Kim, Y.S.; Xu, Z.G.; Lanting, L.; Natarajan, R. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J. Biol. Chem. 2006, 281, 13685–13693. [Google Scholar]
- Riuzzi, F.; Sorci, G.; Donato, R. The amphoterin (HMGB1)/receptor for advanced glycation end products (RAGE) pair modulates myoblast proliferation, apoptosis, adhesiveness, migration, and invasiven. Functional inactivation of RAGE in L6 myoblasts results in tumor formation in vivo. J. Biol. Chem. 2006, 281, 8242–8253. [Google Scholar] [CrossRef]
- Chavakis, E.; Hain, A.; Vinci, M.; Carmona, G.; Bianchi, M.E.; Vajkoczy, P.; Zeiher, A.M.; Chavakis, T.; Dimmeler, S. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ. Res. 2007, 100, 204–212. [Google Scholar] [CrossRef]
- Orlova, V.V.; Choi, E.Y.; Xie, C.; Chavakis, E.; Bierhaus, A.; Ihanus, E.; Ballantyne, C.M.; Gahmberg, C.G.; Bianchi, M.E.; Nawroth, P.P.; et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007, 26, 1129–1139. [Google Scholar] [CrossRef]
- Saleh, A.; Smith, D.R.; Tessler, L.; Mateo, A.R.; Martens, C.; Schartner, E.; van der Ploeg, R.; Toth, C.; Zochodne, D.W.; Fernyhough, P. Receptor for advanced glycation end-products (RAGE) activates divergent signaling pathways to augment neurite outgrowth of adult sensory neurons. Exp. Neurol. 2013, 249, 149–159. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Oppenheim, R.W.; Prevette, D.; Moore, B.W.; Brackenbury, R.; Ratner, N. S100 is present in developing chicken neurons and Schwann cells and promotes motor neuron survival in vivo. J. Neurobiol. 1992, 23, 451–466. [Google Scholar] [CrossRef]
- Haglid, K.G.; Yang, Q.; Hamberger, A.; Bergman, S.; Widerberg, A.; Danielsen, N. S-100β stimulates neurite outgrowth in the rat sciatic nerve grafted with acellular muscle transplants. Brain Res. 1997, 753, 196–201. [Google Scholar] [CrossRef]
- Alexanian, A.R.; Bamburg, J.R. Neuronal survival activity of s100betabeta is enhanced by calcineurin inhibitors and requires activation of NF-κB. FASEB J. 1999, 13, 1611–1620. [Google Scholar]
- Wu, Y.Y.; Bradshaw, R.A. Induction of neurite outgrowth by interleukin-6 is accompanied by activation of Stat3 signaling pathway in a variant PC12 cell (E2) line. J. Biol. Chem. 1996, 271, 13023–13032. [Google Scholar] [CrossRef]
- Wu, C.L.; Chou, Y.H.; Chang, Y.J.; Teng, N.Y.; Hsu, H.L.; Chen, L. Interplay between cell migration and neurite outgrowth determines SH2B1β-enhanced neurite regeneration of differentiated PC12 cells. PLoS One 2012, 7, E34999. [Google Scholar]
- Woszczycka-Korczynska, I.; Olakowska, E.; Marcol, W.; Lewin-Kowalik, J.; Jedrzejowska-Szypulka, H. Schwann cells in therapy of spinal cord injuries. Postepy Higieny i Medycyny Doswiadczalnej 2013, 67, 680–689. [Google Scholar] [CrossRef]
- Ozdemir, M.; Attar, A.; Kuzu, I. Regenerative treatment in spinal cord injury. Curr. Stem Cell Res. Ther. 2012, 7, 364–369. [Google Scholar] [CrossRef]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef]
- Biernaskie, J.; Sparling, J.S.; Liu, J.; Shannon, C.P.; Plemel, J.R.; Xie, Y.; Miller, F.D.; Tetzlaff, W. Skin-derived precursors generate myelinating Schwann cells that promote remyelination and functional recovery after contusion spinal cord injury. J. Neurosci. 2007, 27, 9545–9559. [Google Scholar] [CrossRef]
- Bunge, R.P.; Puckett, W.R.; Becerra, J.L.; Marcillo, A.; Quencer, R.M. Observations on the pathology of human spinal cord injury. A review and classification of 22 new cases with details from a case of chronic cord compression with extensive focal demyelination. Adv. Neurol. 1993, 59, 75–89. [Google Scholar]
- Plemel, J.R.; Duncan, G.; Chen, K.W.; Shannon, C.; Park, S.; Sparling, J.S.; Tetzlaff, W. A graded forceps crush spinal cord injury model in mice. J. Neurotrauma 2008, 25, 350–370. [Google Scholar] [CrossRef]
- Wiliams, R.R.; Bunge, M.B. Schwann cell transplantation: A repair strategy for spinal cord injury? Prog. Brain Res. 2012, 201, 295–312. [Google Scholar] [CrossRef]
- Akassoglou, K.; Yu, W.M.; Akpinar, P.; Strickland, S. Fibrin inhibits peripheral nerve remyelination by regulating Schwann cell differentiation. Neuron 2002, 33, 861–875. [Google Scholar] [CrossRef]
- Rong, L.L.; Trojaborg, W.; Qu, W.; Kostov, K.; Yan, S.D.; Gooch, C.; Szabolcs, M.; Hays, A.P.; Schmidt, A.M. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J. 2004, 18, 1812–1817. [Google Scholar] [CrossRef]
- Rong, L.L.; Yan, S.F.; Wendt, T.; Hans, D.; Pachydaki, S.; Bucciarelli, L.G.; Adebayo, A.; Qu, W.; Lu, Y.; Kostov, K.; et al. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004, 18, 1818–1825. [Google Scholar] [CrossRef]
- Dobrowsky, R.T.; Rouen, S.; Yu, C. Altered neurotrophism in diabetic neuropathy: Spelunking the caves of peripheral nerve. J. Pharmacol. Exp. Ther. 2005, 313, 485–491. [Google Scholar] [CrossRef]
- Perrone, L.; Peluso, G.; Melone, M.A. RAGE recycles at the plasma membrane in S100B secretory vesicles and promotes Schwann cells morphological changes. J. Cell. Physiol. 2008, 217, 60–71. [Google Scholar] [CrossRef]
- Sbai, O.; Devi, T.S.; Melone, M.A.; Feron, F.; Khrestchatisky, M.; Singh, L.P.; Perrone, L. RAGE-TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J. Cell Sci. 2010, 123, 4332–4339. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Song, J.; Lee, W.T.; Park, K.A.; Lee, J.E. Receptor for Advanced Glycation End Products (RAGE) and Its Ligands: Focus on Spinal Cord Injury. Int. J. Mol. Sci. 2014, 15, 13172-13191. https://doi.org/10.3390/ijms150813172
Song J, Lee WT, Park KA, Lee JE. Receptor for Advanced Glycation End Products (RAGE) and Its Ligands: Focus on Spinal Cord Injury. International Journal of Molecular Sciences. 2014; 15(8):13172-13191. https://doi.org/10.3390/ijms150813172
Chicago/Turabian StyleSong, Juhyun, Won Taek Lee, Kyung Ah Park, and Jong Eun Lee. 2014. "Receptor for Advanced Glycation End Products (RAGE) and Its Ligands: Focus on Spinal Cord Injury" International Journal of Molecular Sciences 15, no. 8: 13172-13191. https://doi.org/10.3390/ijms150813172