Water-Soluble Coenzyme Q10 Inhibits Nuclear Translocation of Apoptosis Inducing Factor and Cell Death Caused by Mitochondrial Complex I Inhibition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
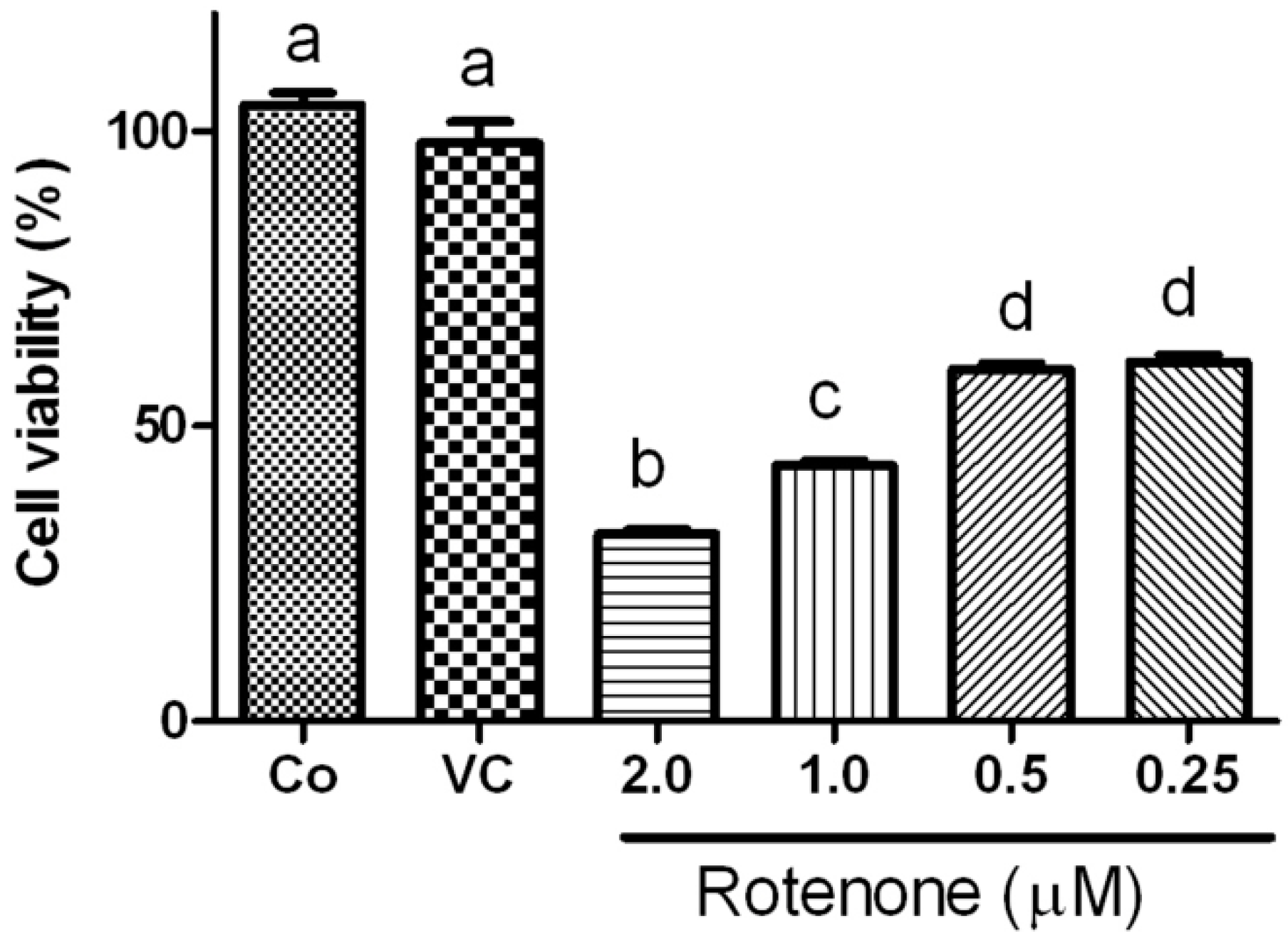
2.1. Effects of Rotenone on Viability of HT22 Cells

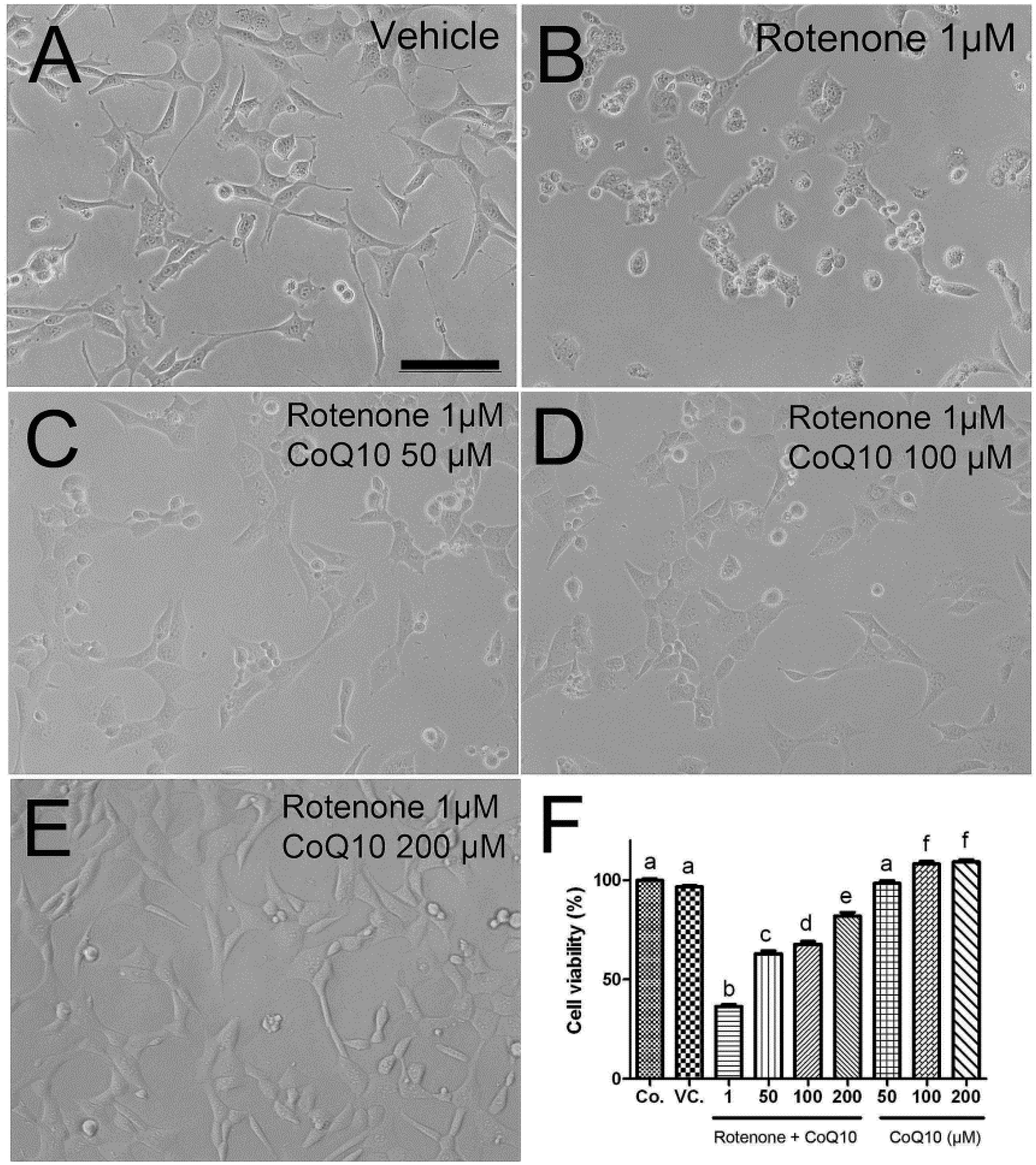
2.2. Protective Effects of CoQ10 against Rotenone Toxicity
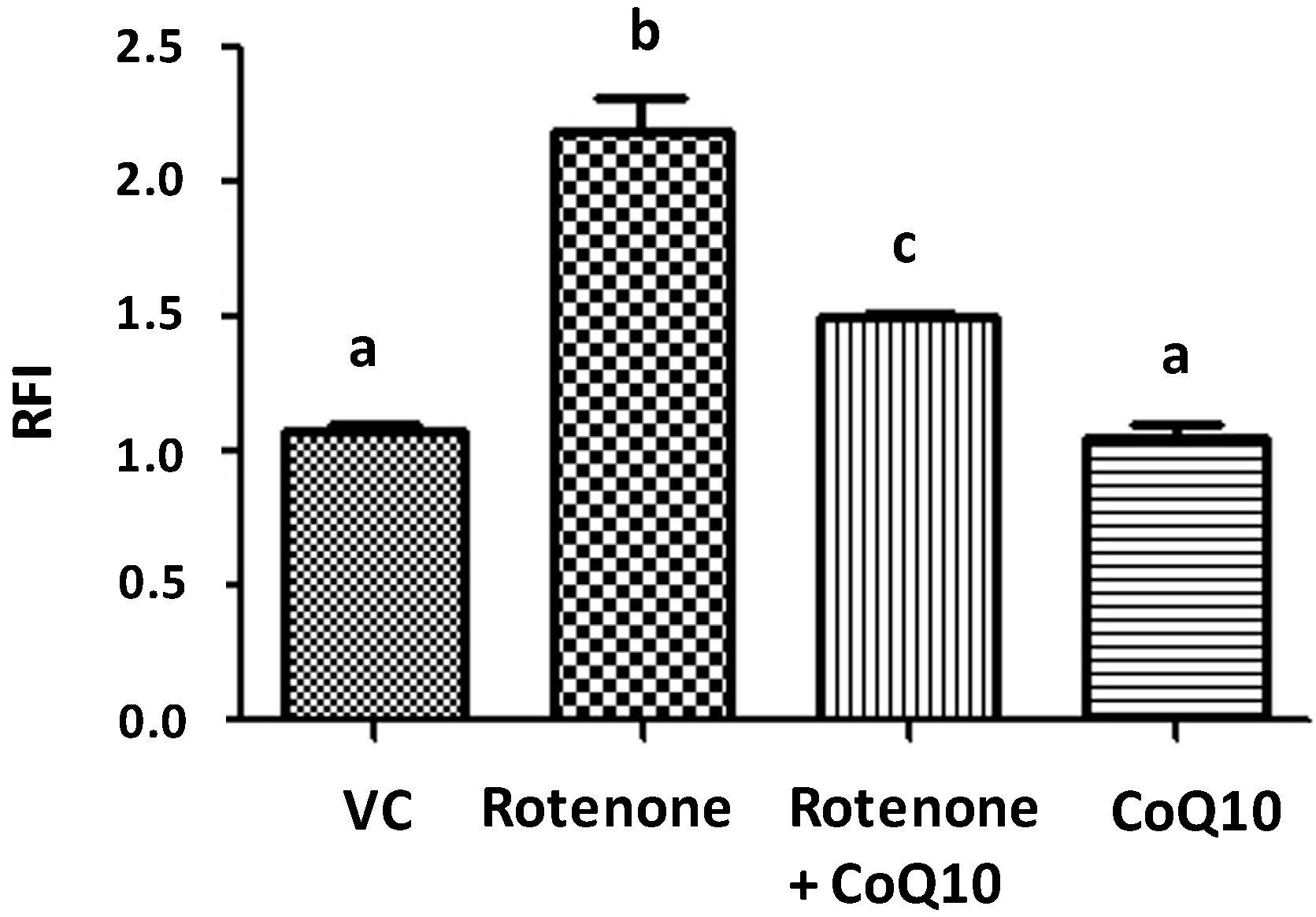
2.3. Effects of CoQ10 on ROS Production


2.4. Effects of CoQ10 on Mitochondrial Membrane Potential

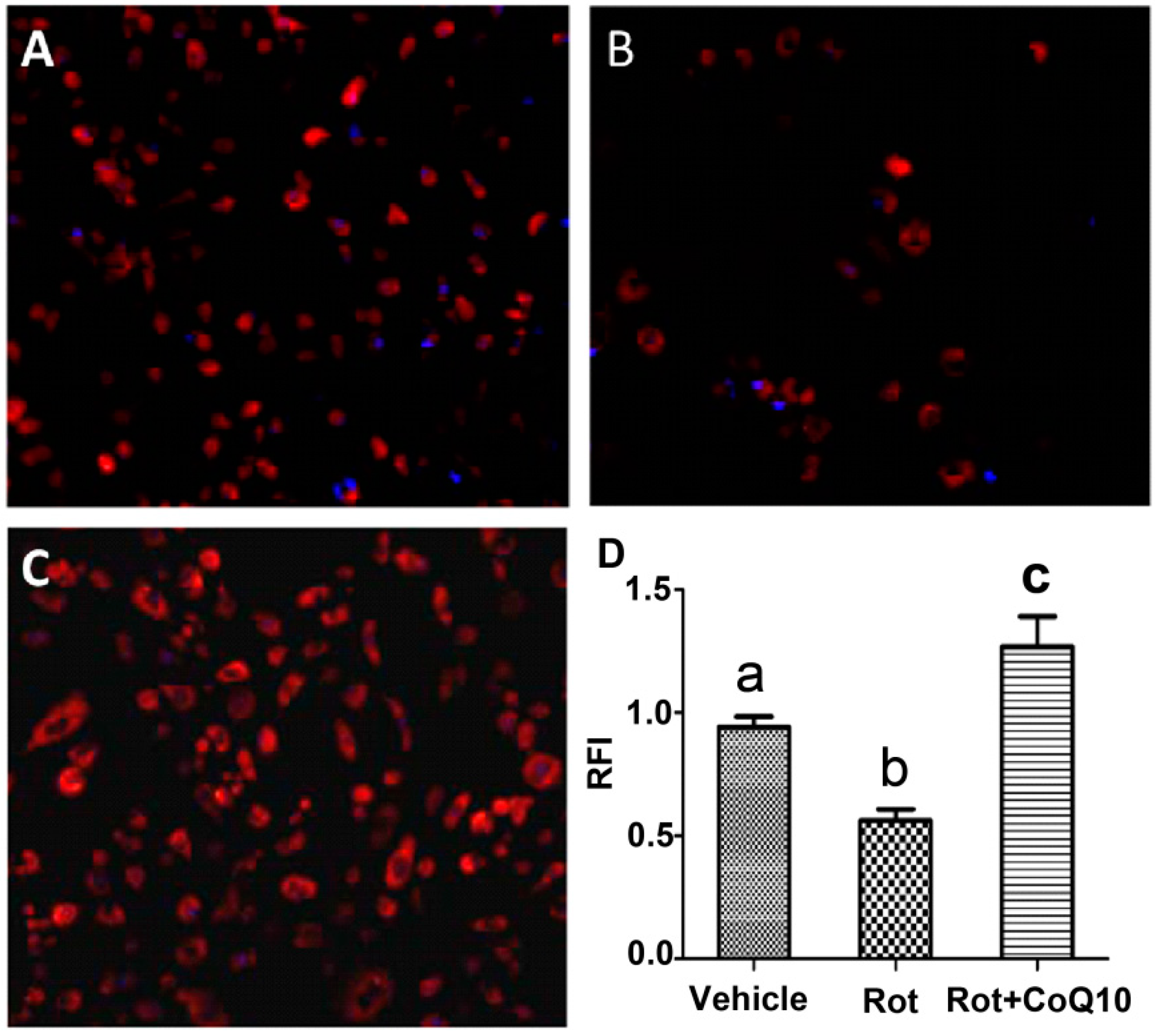
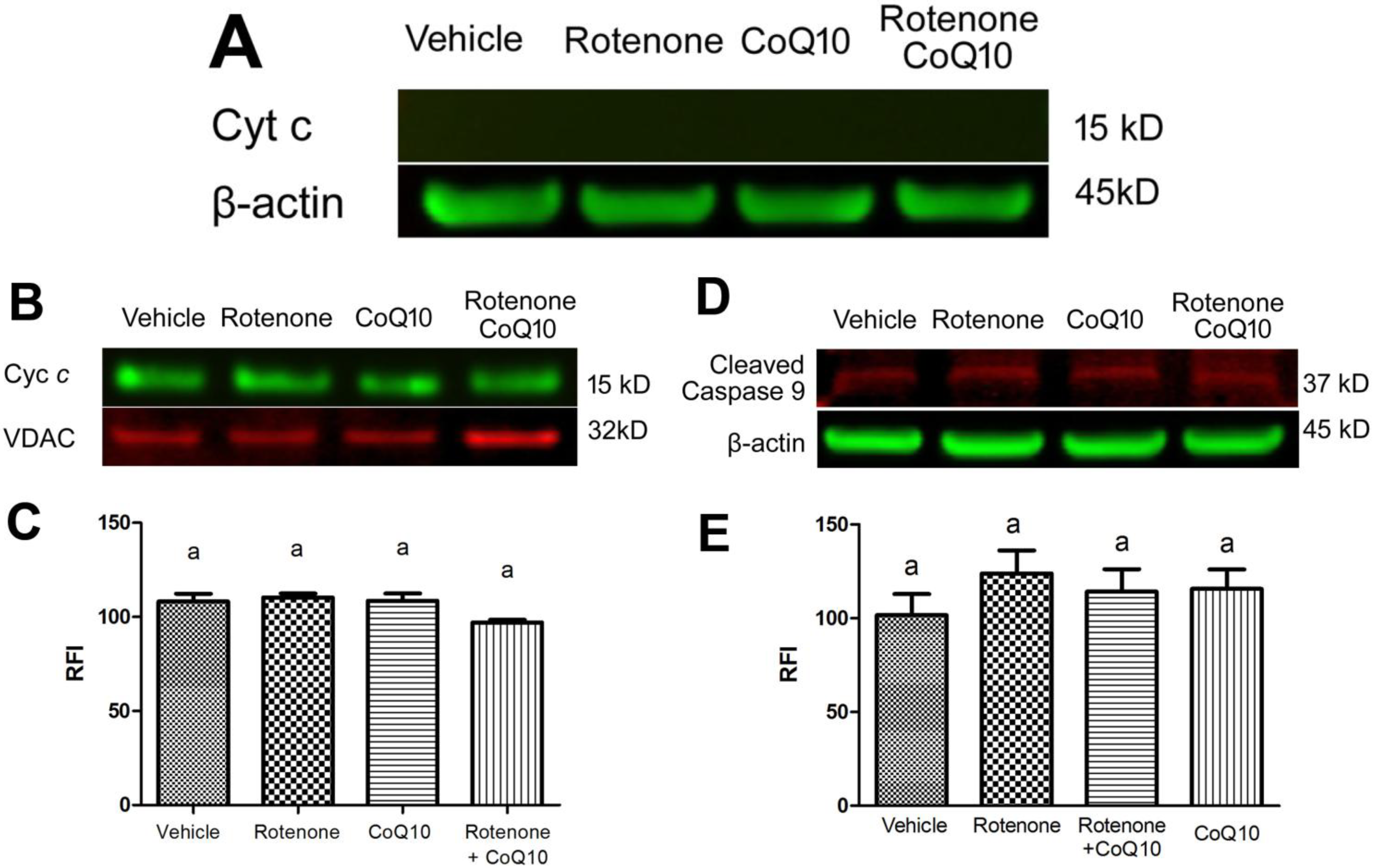
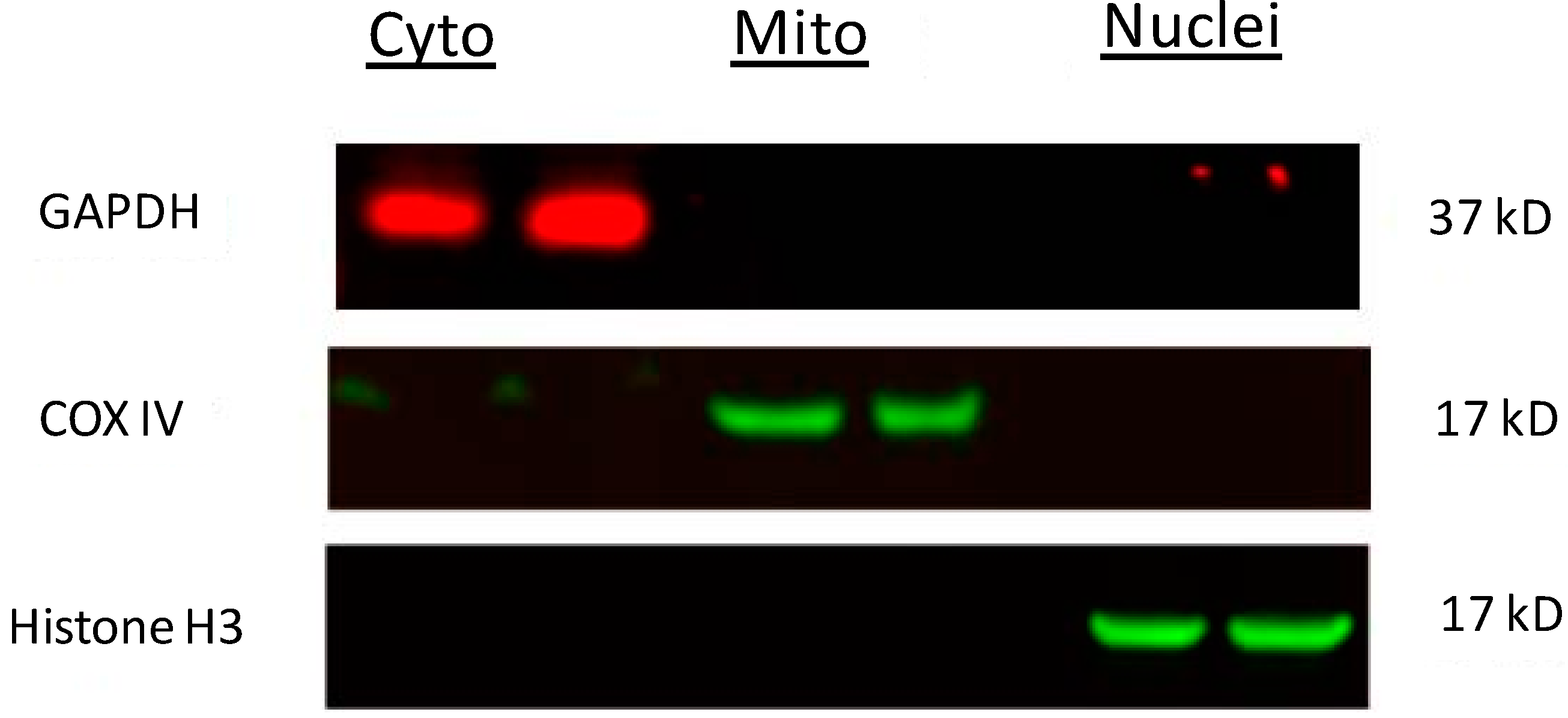
2.5. Blockade of Rotenone-Induced AIF Nuclear Translocation by CoQ10


2.6. Discussion
3. Experimental Section
3.1. Chemicals
3.2. Rotenone and CoQ10 Incubation
3.3. Cell Viability Assay
3.4. Measurements of Superoxide
3.5. Measurements of Mitochondrial Membrane Potential
3.6. Western Blot Analysis

3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in the etiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef]
- Tamilselvam, K.; Braidy, N.; Manivasagam, T.; Essa, M.M.; Prasad, N.R.; Karthikeyan, S.; Thenmozhi, A.J.; Selvaraju, S.; Guillemin, G.J. Neuroprotective effects of hesperidin, a plant flavanone, on rotenone-induced oxidative stress and apoptosis in a celluar model for Parkinson’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 102741. [Google Scholar]
- Radad, K.; Rausch, W.D.; Gille, G. Rotenone induces cell death in primary dopaminergic culture by increasing ROS production and inhibiting mitochondrial respiration. Neurochem. Int. 2006, 49, 379–386. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radial scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef]
- Kooncumcoo, P.; Sharma, S.; Porter, J.; Govitrapong, P.; Ebadi, M. Coenzyme Q10 provides neuroprotection in iron-induced apoptosis in dopaminergic neurons. J. Mol. Neurosci. 2006, 28, 125–141. [Google Scholar] [CrossRef]
- Park, J.; Park, H.H.; Choi, H.; Kim, Y.S.; Yu, H.J.; Lee, Y.J.; Kim, S.H.; Koh, S.H. Coenzyme Q10 protects neural stem cells against hypoxia by enhancing survival signals. Brain Res. 2012, 1478, 64–73. [Google Scholar] [CrossRef]
- Fetoni, A.R.; Piacentini, R.; Fiorita, A.; Paludetti, G.; Troiani, D. Water-soluble coenzyme Q10 formulation (Q-ter) promotes outer hair cell survival in a guinea pig model of noise induced hearing loss (NIHL). Brain Res. 2009, 1537, 108–116. [Google Scholar]
- Panee, J.; Liu, W.; Nakamura, K.; Berry, M.J. The responses of HT 22 cells to the blockade of mitochondrial complexes and potential protective effect of selenium supplementation. Int. J. Biol. Sci. 2007, 3, 335–341. [Google Scholar]
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002, 368, 545–553. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complexes I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Pei, W.; Liou, A.K.; Chen, J. Two caspase-mediated apoptosis pathways induced by rotenone toxicity in cortical neuronal cells. FASEB J. 2003, 17, 520–522. [Google Scholar]
- Green, D.R.; Reed, J.C. Mitochondrial and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef]
- Lee, J.H.; Won, Y.S.; Park, K.H.; Lee, M.K.; Tachibana, H.; Yamada, K.; Seo, K.I. Celastrol inhibits growth and induces apoptosis cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis 2012, 17, 1275–1286. [Google Scholar] [CrossRef]
- Lin, T.K.; Cheng, C.H.; Chen, S.D.; Liou, C.W.; Huang, C.R.; Chuang, Y.C. Mitochondrial dysfunction and oxidative stress promote apoptotic cell death in the striatum via cytochrome c/caspase-3 signaling cascade following chronic rotenone intoxication in rats. Int. J. Mol. Sci. 2012, 13, 8722–8739. [Google Scholar] [CrossRef]
- Büttner, S.; Habernig, L.; Broeskamp, F.; Ruli, D.; Vögtle, F.N.; Vlachos, M.; Macchi, F.; Küttner, V.; Carmona-Gutierrez, D.; Eisenberg, T.; et al. Endonuclease G mediates α-synuclein cytotoxicity during Parkinson’s disease. EMBO J. 2013, 32, 3041–3054. [Google Scholar] [CrossRef] [Green Version]
- Krantic, S.; Mechawar, N.; Reix, S.; Quirion, R. Apoptosis-inducing factor: A matter of neuron life and death. Prog. Neurobiol. 2007, 81, 179–196. [Google Scholar] [CrossRef]
- Li, J.; Spletter, M.L.; Johnson, D.A.; Wright, L.S.; Svendsen, C.N.; Johnson, J.A. Rotenone-induced caspase 9/3-independent and -dependent cell death in undifferentiated human neural stem cells. J. Neurochem. 2005, 92, 462–476. [Google Scholar] [CrossRef]
- Liang, J.M.; Yu, Y.; Wang, B.Y.; Lu, B.; Zhang, J.Z.; Zhang, H.B.; Ge, P.F. Ginsenoside Rb1 attenuates oxygen-glucose deprivation-induced apoptosis in SH-SY5Y cells via protection of mitochondrial and inhibition of AIF and cytochrome C release. Molecules 2013, 18, 12777–12792. [Google Scholar] [CrossRef]
- Shih, C.M.; Ko, W.C.; Wu, J.S.; Wei, Y.H.; Wang, L.F.; Cheng, H.H.; Chen, C.T. Mediating of caspase-independent apoptosis by cadmium through the mitochondrial-ROS pathway in MRC-5 fibroblasts. J. Cell. Biochem. 2004, 91, 384–397. [Google Scholar] [CrossRef]
- Chinta, S.J.; Rane, A.; Yadava, N.; Andersen, J.; Nicholls, D.G.; Polster, B.M. Reactive oxygen species regulation by AIF-and complex I-depleted brain mitochondria. Free Radic. Biol. Med. 2009, 46, 939–947. [Google Scholar] [CrossRef]
- Moon, Y.; Lee, K.H.; Park, J.-H.; Geum, D.; Kim, K. Mitochondrial membrane depolarization and the selective death of dopaminergic neurons by rotenone: Protective effect of coenzyme Q10. J. Neurochem. 2005, 93, 1199–11208. [Google Scholar] [CrossRef]
- Sandhu, J.K.; Pandey, S.; Ribecco-Lutkiewicz, M.; Monette, R.; Borowy-Borowsdi, H.; Walker, P.R.; Sikorska, M. Molecular mechanisms of glutamate neurotoxicity in mixed cultures of NT2-derived neurons and astrocytes: Protective effects of Coenzyme Q10. J. Neurosci. Res. 2003, 72, 691–703. [Google Scholar] [CrossRef]
- Jing, L.; Kumari, S.; Mendelev, N.; Li, P.A. Coenzyme Q10 ameliorates ultraviolet B irradiation induced cell death through inhibition of mitochondrial intrinsic cell death pathway. Int. J. Mol. Sci. 2011, 12, 8302–8315. [Google Scholar] [CrossRef]
- Somayajulu-Nitu, M.; Sandhu, J.K.; Cohen, J.; Sikorska, M.; Sridhar, T.S.; Matei, A.; Borowy-Borowski, H.; Pandey, S. Paraquat induces oxidative stress, neuronal loss in substantia nigra region and Parkinsonism in adult rats: Neuroprotection and amelioration of symptoms by water-soluble formulation of Coenzyme Q10. BMC Neurosci. 2009, 10, 1–12. [Google Scholar] [CrossRef]
- Mendelev, N.; Mehta, S.L.; Witherspoon, S.; He, Q.; Sexton, J.Z.; Li, P.A. Upregulation of human selenoprotein H in murine hippocampal neuronal cells promotes mitochondrial biogenesis and functional performance. Mitochondrion 2011, 11, 76–82. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, H.; Chen, G.; Ma, W.; Li, P.-A.A. Water-Soluble Coenzyme Q10 Inhibits Nuclear Translocation of Apoptosis Inducing Factor and Cell Death Caused by Mitochondrial Complex I Inhibition. Int. J. Mol. Sci. 2014, 15, 13388-13400. https://doi.org/10.3390/ijms150813388
Li H, Chen G, Ma W, Li P-AA. Water-Soluble Coenzyme Q10 Inhibits Nuclear Translocation of Apoptosis Inducing Factor and Cell Death Caused by Mitochondrial Complex I Inhibition. International Journal of Molecular Sciences. 2014; 15(8):13388-13400. https://doi.org/10.3390/ijms150813388
Chicago/Turabian StyleLi, Haining, Guisheng Chen, Wanrui Ma, and Ping-An Andy Li. 2014. "Water-Soluble Coenzyme Q10 Inhibits Nuclear Translocation of Apoptosis Inducing Factor and Cell Death Caused by Mitochondrial Complex I Inhibition" International Journal of Molecular Sciences 15, no. 8: 13388-13400. https://doi.org/10.3390/ijms150813388