Theoretical Study of the Transpore Velocity Control of Single-Stranded DNA

Abstract
:1. Introduction

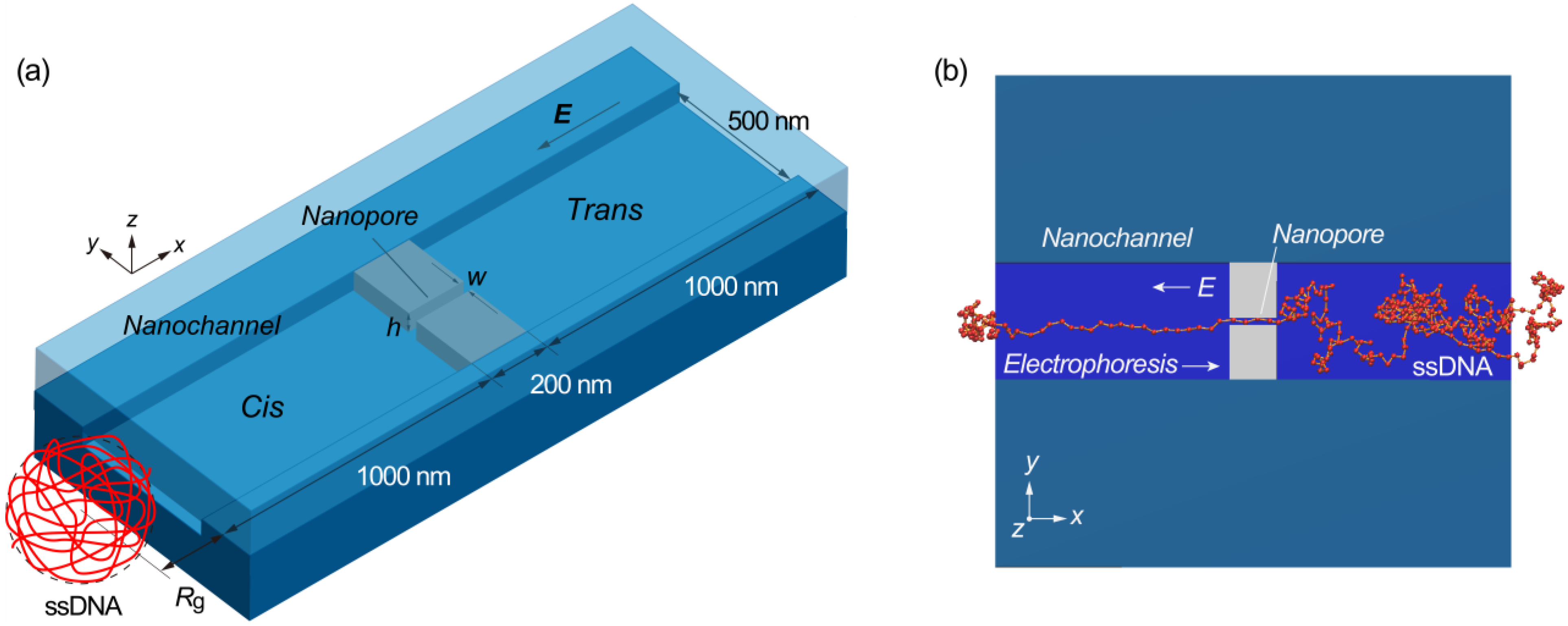{kind=link}
{kind=link}
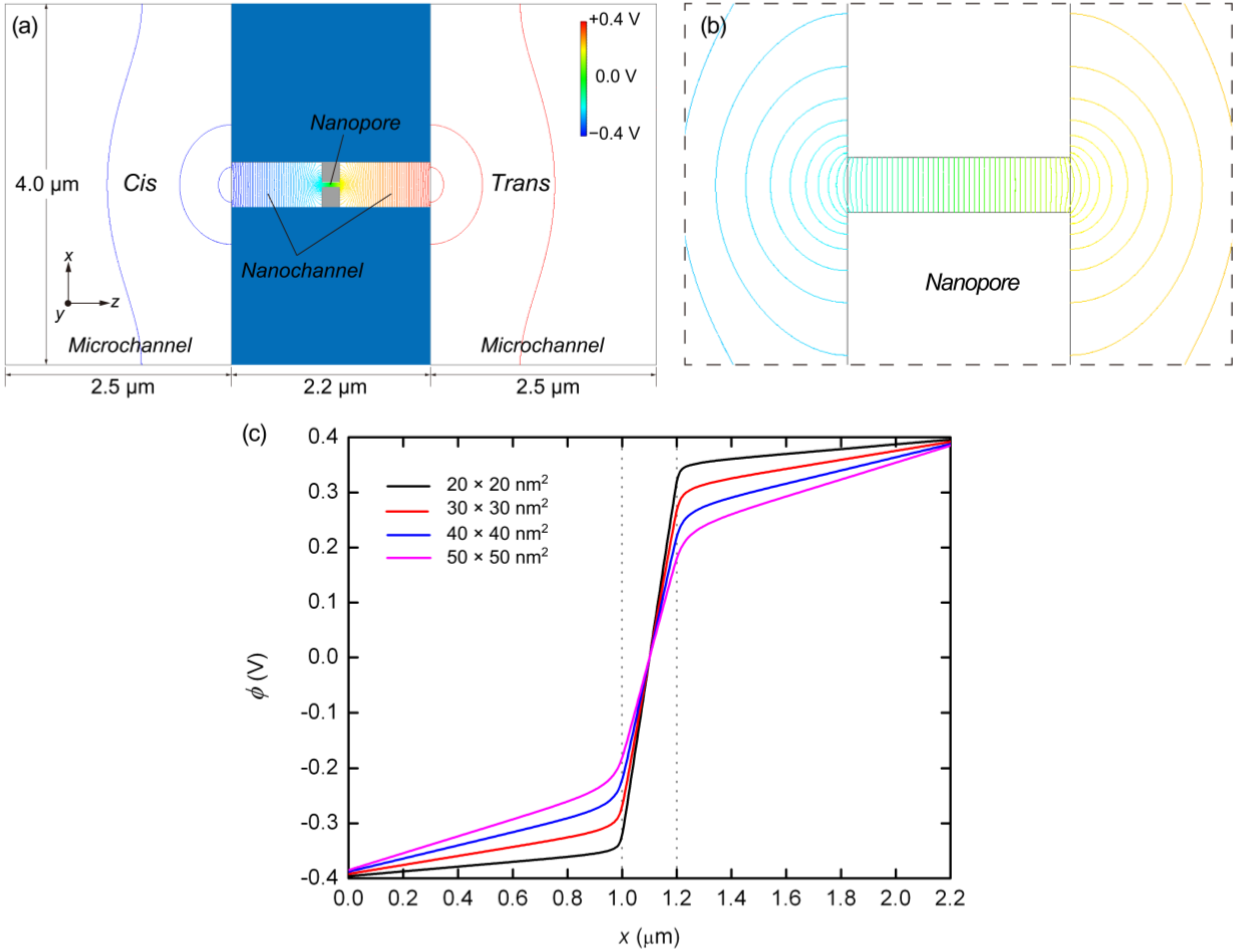{kind=link}
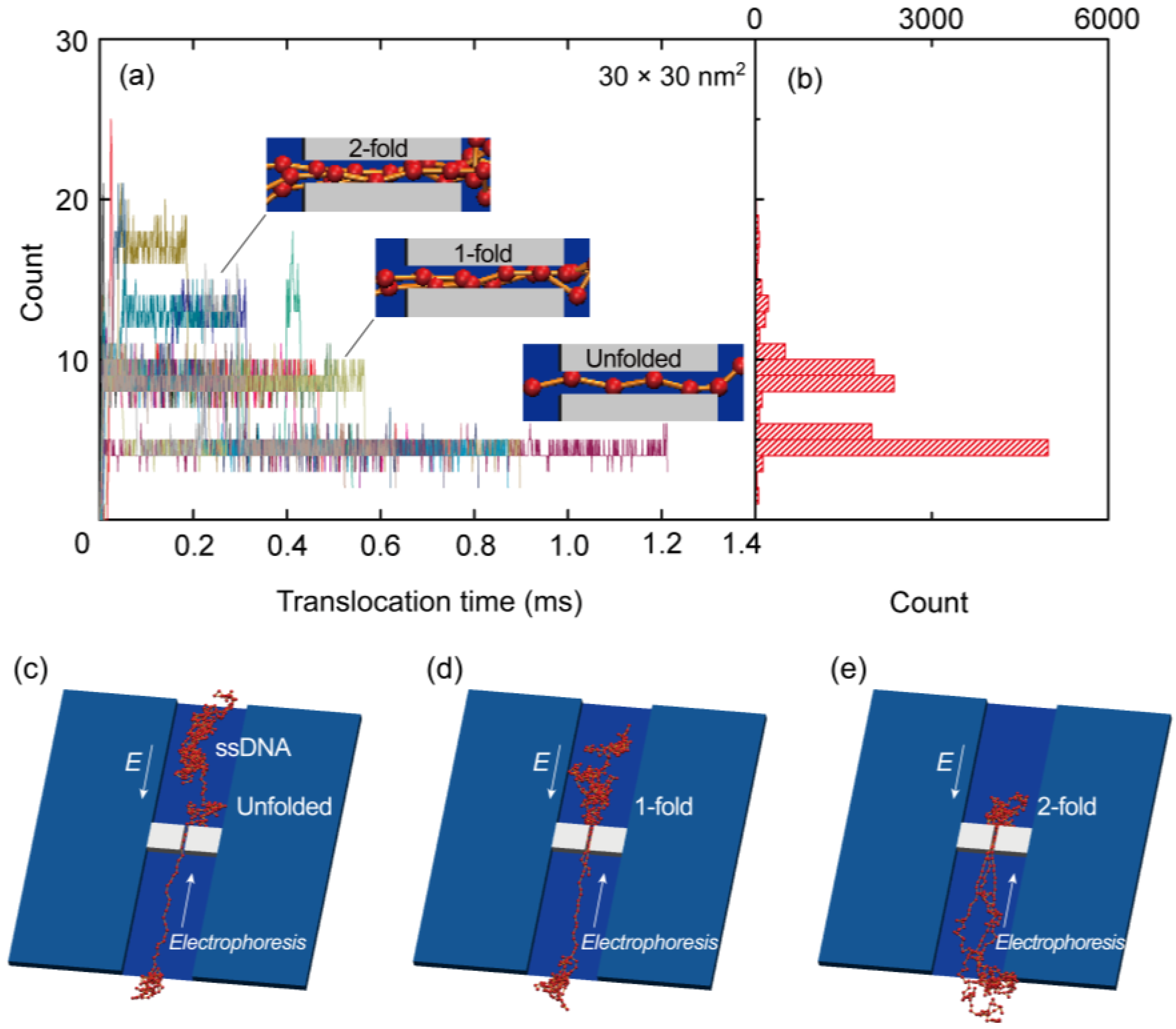{kind=link}
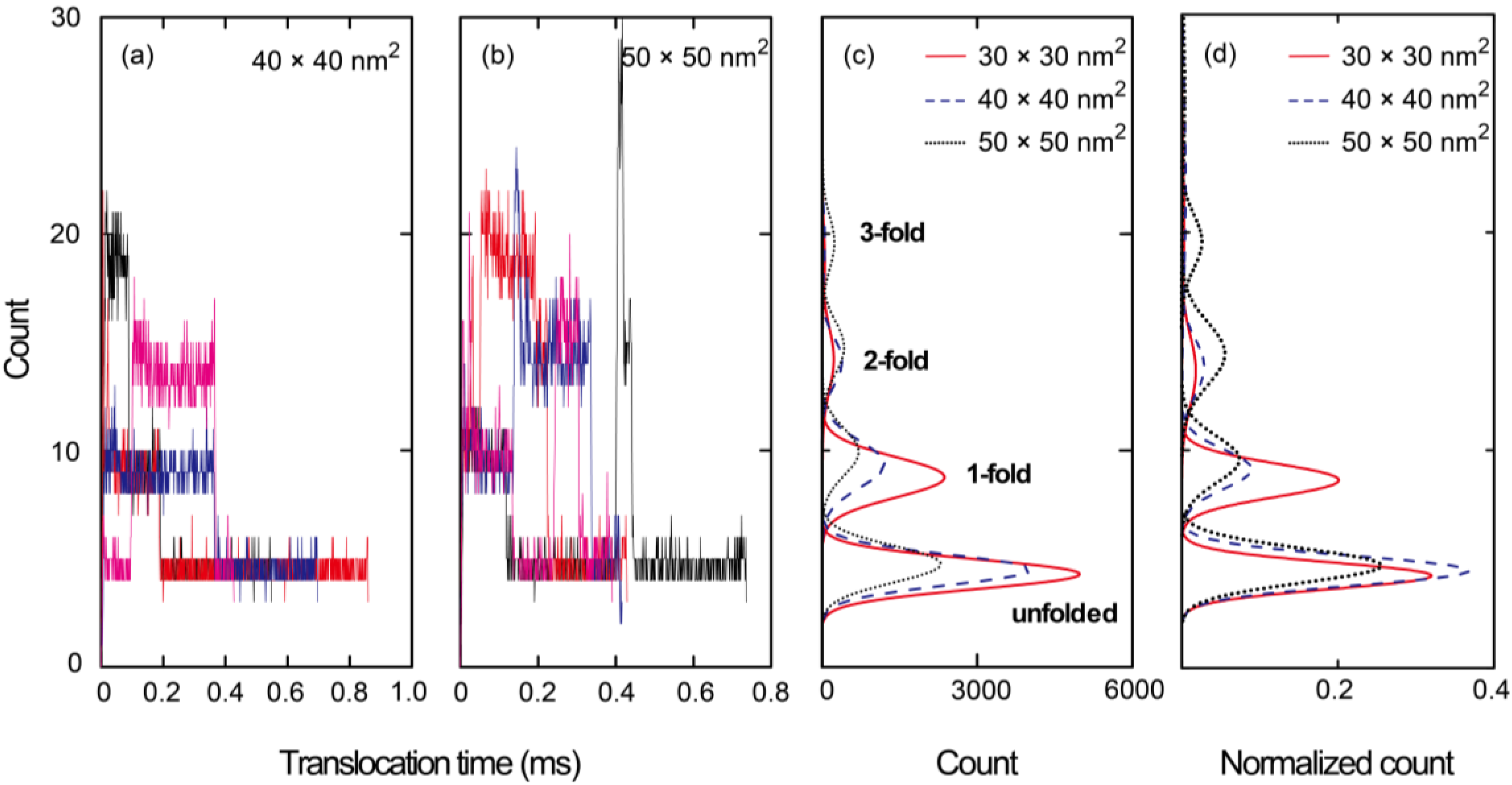{kind=link}
{kind=link}
| Group | Pore Type | Diameter (nm) | Length (nm) | Voltage (mV) | Voltage/Length (×106 V/m) | Polymer Length (bp or nt) | Polymer Type |
|---|---|---|---|---|---|---|---|
| Kasianowicz et al. [5] | α-HL | 1.3 | 5.2 | 120 | 23 | 150 | ssDNA, ssRNA * |
| Meller et al. [18,19,20] | α-HL | 1.3 | 5.2 | 50–300 | 9.6–58 | 5–100 | ssDNA |
| Butler et al. [35] | MspA | 1 | 10 | 140, 180 | 14, 18 | 50 | ssDNA |
| Wendell et al. [36] | Phi29 | 3.3 | 7.5 | 40, 75 | 5.3, 10 | 5.5 k | dsDNA |
| Franceschini et al. [37] | ClyA | 7.8 | 13 | 100 | 7.7 | 290 bp, 51 nt | dsDNA, ssDNA |
| Li et al. [21] | SiN | 3, 10 | 5–10 | 60, 120 | 6–24 | 3–10 k | dsDNA |
| Storm et al. [22] | SiN | 10 | 20 | 100–600 | 5–30 | 10–97 k | dsDNA |
| Skinner et al. [24] | SiN | 10 | 20 | 100–600 | 5–30 | 10–30 k | dsDNA, dsRNA, ssRNA * |
| Tsutsui et al. [26] | SiN | 50 | 200 | 1000 | 5 | 48.5 k | dsDNA |
| Fologea et al. [27,28] | SiN | 10 | 10, 280 | 120 | 0.43, 12 | 3 k | dsDNA, ssDNA |
| Schneider et al. [25] | Graphene | 22 | 0.3 | 200 | 670 | 48.5 k | dsDNA |

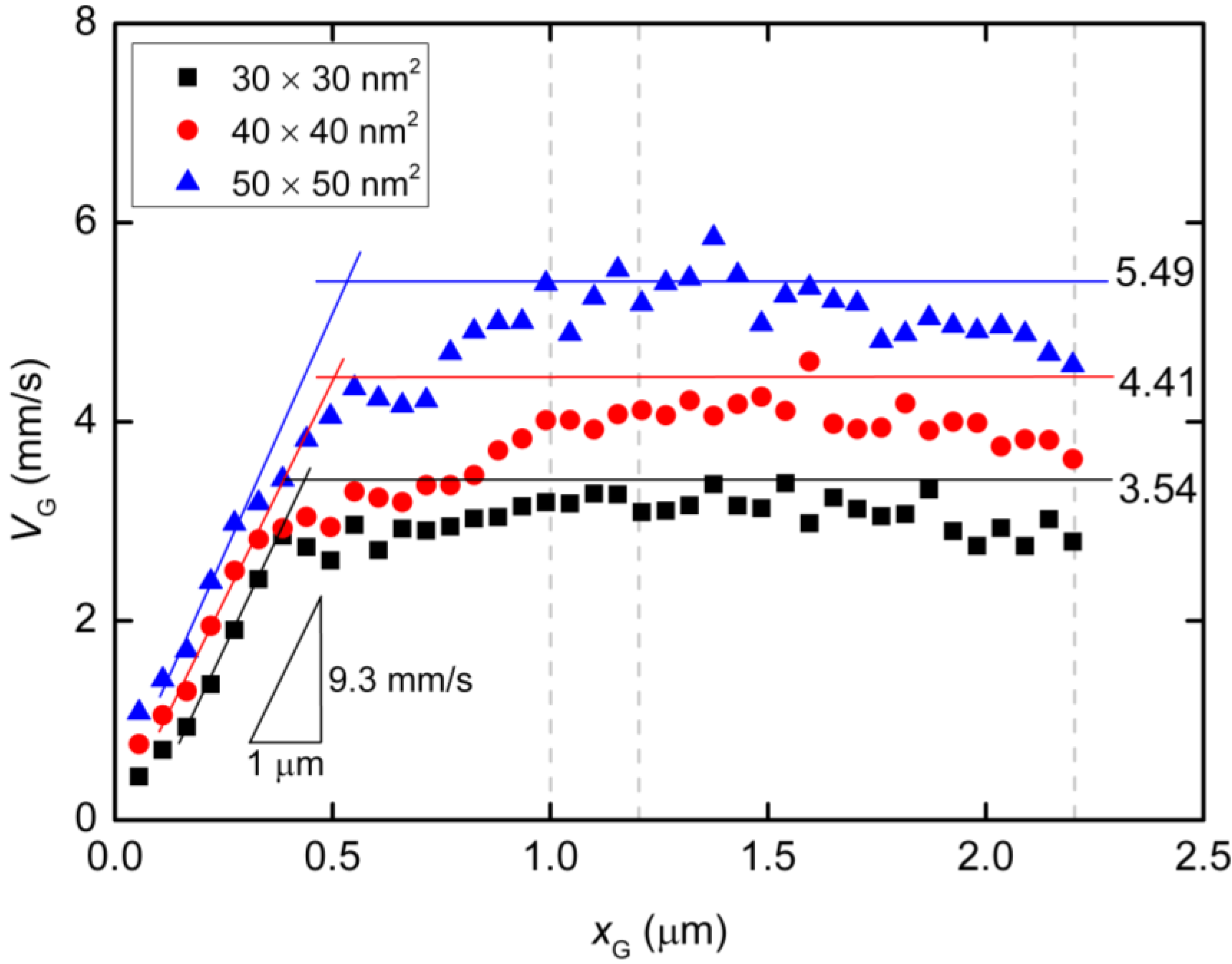
2. Results and Discussion
2.1. Validation of the Coarse-Grained Single-Stranded DNA (ssDNA) Model

2.2. Langevin Dynamics Simulations
| Pore Size (nm2) | Echannel (V/m) | Epore (V/m) | Npore | ΔN/Δx (m−1) | vchannel (mm/s) | vpore (mm/s) |
|---|---|---|---|---|---|---|
| 30 × 30 | 8.2 × 104 | 2.7 × 106 | 6.48 | 1.30 × 109 | 2.3 | 1.3 |
| 40 × 40 | 1.2 × 105 | 2.2 × 106 | 6.81 | 9.64 × 108 | 3.3 | 1.1 |
| 50 × 50 | 1.5 × 105 | 1.9 × 106 | 9.29 | 7.45 × 108 | 4.3 | 1.2 |









3. Langevin Dynamics Simulations of Polymer Chain Motion



4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dekker, C. Solid-state nanopores. Nat. Nanotechnol. 2007, 2, 209–215. [Google Scholar] [CrossRef]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X.; et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 2008, 26, 1146–1153. [Google Scholar] [CrossRef]
- Zwolak, M.; di Ventra, M. Colloquium: Physical approaches to DNA sequencing and detection. Rev. Mod. Phys. 2008, 80, 141–165. [Google Scholar]
- Venkatesan, B.M.; Bashir, R. Nanopore sensors for nucleic acid analysis. Nat. Nanotechnol. 2011, 6, 615–624. [Google Scholar]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef]
- Clarke, J.; Wu, H.-C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef]
- Lieberman, K.R.; Cherf, G.M.; Doody, M.J.; Olasagasti, F.; Kolodji, Y.; Akeson, M. Processive replication of single DNA molecules in a nanopore catalyzed by phi29 DNA polymerase. J. Am. Chem. Soc. 2010, 132, 17961–17972. [Google Scholar]
- Lagerqvist, J.; Zwolak, M.; di Ventra, M. Fast DNA sequencing via transverse electronic transport. Nano Lett. 2006, 6, 779–782. [Google Scholar]
- Liang, X.; Chou, S.Y. Nanogap detector inside nanofluidic channel for fast real-time label-free DNA analysis. Nano Lett. 2008, 8, 1472–1476. [Google Scholar] [CrossRef]
- Tsutsui, M.; Taniguchi, M.; Yokota, K.; Kawai, T. Identifying single nucleotides by tunnelling current. Nat. Nanotechnol. 2010, 5, 286–290. [Google Scholar] [CrossRef]
- Szarek, P.; Suwannawong, S.; Doi, K.; Kawano, S. Theoretical study on physicochemical aspects of a single molecular junction: Application to the bases of ssDNA. J. Phys. Chem. C 2013, 117, 109809:1–109809:9. [Google Scholar]
- Dorfman, K.D. DNA electrophoresis in microfabricated devices. Rev. Mod. Phys. 2010, 82, 2903–2947. [Google Scholar] [CrossRef]
- Sung, W.; Park, P.J. Polymer translocation through a pore in a membrane. Phys. Rev. Lett. 1996, 77, 783–786. [Google Scholar] [CrossRef]
- Muthukumar, M. Polymer translocation through a hole. J. Chem. Phys. 1999, 111, 10371–10374. [Google Scholar]
- Huopaniemi, I.; Luo, K.; Ala-Nissila, T.; Ying, S.-C. Langevin dynamics simulations of polymer translocation through nanopores. J. Chem. Phys. 2006, 125, 124901:1–124901:8. [Google Scholar]
- Nagahiro, S.; Kawano, S.; Kotera, H. Separation of long DNA chains using a nonuniform electric field: A numerical study. Phys. Rev. E 2007, 75, 011902:1–011902:5. [Google Scholar]
- Uehara, S.; Tsutsui, M.; Doi, K.; Taniguchi, M.; Kawano, S.; Kawai, T. Fluid dynamics and electrical detection of λDNA in electrode-embedded nanochannels. J. Biomech. Sci. Eng. 2013, 8, 244–256. [Google Scholar] [CrossRef]
- Meller, A.; Nivon, L.; Brandin, E.; Golovchenko, J.; Branton, D. Rapid nanopore discrimination between single polynucleotide molecules. Proc. Natl. Acad. Sci. USA 2000, 97, 1079–1084. [Google Scholar]
- Meller, A.; Nivon, L.; Branton, D. Voltage-driven DNA translocations through a nanopore. Phys. Rev. Lett. 2001, 86, 3435–3438. [Google Scholar] [CrossRef]
- Meller, A.; Branton, D. Single molecule measurements of DNA transport through a nanopore. Electrophoresis 2002, 23, 2583–2591. [Google Scholar] [CrossRef]
- Li, J.; Gershow, M.; Stein, D.; Brandin, E.; Golovchenko, J.A. DNA molecules and configurations in a solid-state nanopore microscope. Nat. Mater. 2003, 2, 611–615. [Google Scholar]
- Storm, A.J.; Storm, C.; Chen, J.; Zandbergen, H.; Joanny, J.-F.; Dekker, C. Fast DNA translocation through a solid-state nanopore. Nano Lett. 2005, 5, 1193–1197. [Google Scholar] [CrossRef]
- Krasilnikov, O.V.; Rodrigues, C.G.; Bezrukov, S.M. Single polymer molecules in a protein nanopore in the limit of a strong polymer-pore attraction. Phys. Rev. Lett. 2006, 97, 018301:1–018301:4. [Google Scholar]
- Skinner, G.M.; van den Hout, M.; Broekmans, O.; Dekker, C.; Dekker, N.H. Distinguishing single- and double-stranded nucleic acid molecules using solid-state nanopores. Nano Lett. 2009, 9, 2953–2960. [Google Scholar] [CrossRef]
- Schneider, G.F.; Kowalczyk, S.W.; Calado, V.E.; Pandraud, G.; Zandbergen, H.W.; Vandersypen, L.M.K.; Dekker, C. DNA translocation through graphene nanopores. Nano Lett. 2010, 10, 3163–3167. [Google Scholar] [CrossRef]
- Tsutsui, M.; He, Y.; Furuhashi, M.; Rahong, S.; Taniguchi, M.; Kawai, T. Transverse electric field dragging of DNA in a nanochannel. Sci. Rep. 2012, 2, 394. [Google Scholar]
- Fologea, D.; Uplinger, J.; Thomas, B.; McNabb, D.S.; Li, J. Slowing DNA translocation in a solid-state nanopore. Nano Lett. 2005, 5, 1734–1737. [Google Scholar] [CrossRef]
- Fologea, D.; Gershow, M.; Ledden, B.; McNabb, D.S.; Golovchenko, J.A.; Li, J. Detecting single stranded DNA with a solid state nanopore. Nano Lett. 2005, 5, 1905–1909. [Google Scholar] [CrossRef]
- Lehtola, V.V.; Linna, R.P.; Kaski, K. Dynamics of forced biopolymer translocation. EPL 2009, 85, 58006:1–58006:6. [Google Scholar]
- Yong, H.; Wang, Y.; Yuan, S.; Xu, B.; Luo, K. Driven polymer translocation through a cylindrical nanochannel: Interplay between the channel length and the chain length. Soft Matter 2012, 8, 2769–2774. [Google Scholar] [CrossRef]
- Edmonds, C.M.; Hudiono, Y.C.; Ahmadi, A.G.; Hesketh, P.J.; Nair, S. Polymer translocation in solid-state nanopores: Dependence of scaling behavior on pore dimensions and applied voltage. AIP 2012, 136, 065105:1–065105:10. [Google Scholar]
- Luo, K.; Ala-Nissila, T.; Ying, S.-C.; Bhattacharya, A. Sequence dependence of DNA translocation through a nanopore. Phys. Rev. Lett. 2008, 100, 058101:1–058101:4. [Google Scholar]
- Ramachandran, A.; Guo, Q.; Iqbal, S.M.; Liu, Y. Coarse-grained molecular dynamics simulation of DNA translocation in chemically modified nanopores. J. Phys. Chem. B 2011, 115, 6138–6148. [Google Scholar]
- Forrey, C.; Muthukumar, M. Langevin dynamics simulations of dsDNA translocation through synthetic nanopores. J. Chem. Phys. 2007, 127, 015102:1–015102:10. [Google Scholar]
- Butler, T.Z.; Pavlenok, M.; Derrington, I.M.; Niederweis, M.; Gundlach, J.H. Single-molecule DNA detection with an engineered MspA protein nanopore. Proc. Natl. Acad. Sci. USA 2008, 105, 20647–20652. [Google Scholar]
- Wendell, D.; Jing, P.; Geng, J.; Subramaniam, V.; Lee, T.J.; Montemagnom, C.; Guo, P. Translocation of double stranded DNA through membrane adapted phi29 motor protein nanopore. Nat. Nanotechnol. 2009, 4, 765–772. [Google Scholar] [CrossRef]
- Franceschini, L.; Soskine, M.; Biesemans, A.; Maglia, G. A nanopore machine promotes the vectorial transport of DNA cross membranes. Nat. Commun. 2013, 4, 2415. [Google Scholar]
- Gauthier, M.G.; Slater, G.W. Molecular dynamics simulation of a polymer chain translocating through a nanoscopic pore. Eur. Phys. J. E 2008, 25, 17–23. [Google Scholar] [CrossRef]
- Izmitli, A.; Schwartz, D.C.; Graham, M.D.; de Pablo, J.J. The effect of hydrodynamic interactions on the dynamics of DNA translocation through pores. J. Chem. Phys. 2008, 128, 085102:1–085102:7. [Google Scholar]
- Fyta, M.; Melchionna, S.; Succi, S.; Kaxiras, E. Hydrodynamic correlations in the translocation of a biopolymer through a nanopore: Theory and multiscale simulations. Phys. Rev. E 2008, 78, 036704:1–036704:7. [Google Scholar]
- Luo, K.; Metzler, R. The chain sucker: Translocation dynamics of a polymer chain into a long narrow channel driven by longitudinal flow. J. Chem. Phys. 2011, 134, 135102:1–135102:8. [Google Scholar]
- Kawaguchi, C.; Noda, T.; Tsutsui, M.; Taniguchi, M.; Kawano, S.; Kawai, T. Electrical detection of single pollen allergen particles using electrode-embedded microchannels. J. Phys. 2012, 24, 164202:1–164202:6. [Google Scholar]
- Yasui., T.; Rahong, S.; Motoyama, K.; Yanagida, T.; Wu, Q.; Kaji, N.; Kanai, M.; Doi, K.; Nagashima, K.; Tokeshi, M.; et al. DNA manipulation and separation in sublithographic-scale nanowire array. ACS Nano 2013, 7, 3029–3035. [Google Scholar] [CrossRef]
- He, Y.; Tsutsui, M.; Fan, C.; Taniguchi, M.; Kawai, T. Controlling DNA translocation through gate modulation of nanopore wall surface charges. ACS Nano 2011, 5, 5509–5518. [Google Scholar] [CrossRef]
- Doi, K.; Haga, T.; Shintaku, H.; Kawano, S. Development of coarse-graining DNA models for single-nucleotide resolution analysis. Philos. Trans. R. Soc. A 2010, 368, 2615–2628. [Google Scholar] [CrossRef]
- Doi, K.; Qian, W.; Uehara, S.; Tsutsui, M.; Taniguchi, M.; Kawai, T.; Kawano, S. Langevin dynamics study on electrokinetic transport of long-chained DNA through nanogap embedded in nanochannel. Int. J. Emerg. Multidiscip. Fluid Sci. 2014, in press. [Google Scholar]
- Hanasaki, I.; Takahashi, H.; Sazaki, G.; Nakajima, K.; Kawano, S. Single-molecule measurements and dynamical simulations of protein molecules near silicon substrates. J. Phys. D 2008, 41, 095301:1–095301:9. [Google Scholar]
- Hanasaki, I.; Shintaku, H.; Matsunami, S.; Kawano, S. Structural and tensile properties of self-assembled DNA network on mica surface. Comput. Model. Eng. Sci. 2009, 46, 191–207. [Google Scholar]
- Doi, K.; Uemura, T.; Kawano, S. Molecular dynamics study of solvation effect on diffusivity changes of DNA fragments. J. Mol. Model. 2011, 17, 1457–1465. [Google Scholar]
- Doi, K.; Toyokita, Y.; Akamatsu, S.; Kawano, S. Reaction–diffusion wave model for self-assembled network formation of poly(dA)·poly(dT) DNA on mica and HOPG surfaces. Compt. Methods Biomech. Biomed. Eng. 2014, 17, 661–677. [Google Scholar] [CrossRef]
- Doi, K.; Takeuchi, H.; Nii, R.; Akamatsu, S.; Kakizaki, T.; Kawano, S. Self-assembly of 50 bp poly(dA)·poly(dT) DNA on highly oriented pyrolytic graphite via atomic force microscopy observation and molecular dynamics simulation. J. Chem. Phys. 2013, 139, 085102:1–085102:9. [Google Scholar]
- Stellwagen, E.; Lu, Y.; Stellwagen, N.C. Unified description of electrophoresis and diffusion for DNA and other polyions. Biochemistry 2003, 42, 11745–11750. [Google Scholar] [CrossRef]
- Femtet®; Murata Software Co., Ltd.: Tokyo, Japan, 2013.
- He, Y.; Tsutsui, M.; Fan, C.; Taniguchi, M.; Kawai, T. Gate manipulation of DNA capture into nanopores. ACS Nano 2011, 5, 8391–8397. [Google Scholar] [CrossRef]
- Trepargnier, E.H.; Radenovic, A.; Sivak, D.; Geissler, P.; Liphardt, J. Controlling DNA capture and propagation through artificial nanopores. Nano Lett. 2007, 7, 2824–2830. [Google Scholar] [CrossRef]
- Mannion, J.T.; Reccius, C.H.; Cross, J.D.; Craighead, H.G. Conformational analysis of single DNA molecules undergoing entropically induced motion in nanochannels. Biophys. J. 2006, 90, 4538–4545. [Google Scholar] [CrossRef]
- Salieb-Beugelaar, G.B.; Dorfman, K.D.; van den Berg, A.; Eijkel, J.C.T. Electrophoretic separation of DNA in gels and nanostructures. Lab. Chip 2009, 9, 2508–2523. [Google Scholar] [CrossRef]
- Uehara, S.; Shintaku, H.; Kawano, S. Electrokinetic flow dynamics of weakly aggregated λDNA confined in nanochannels. Trans. ASME J. Fluids Eng. 2011, 133, 121203:1–121203:8. [Google Scholar]
- Tinland, B.; Pluen, A.; Sturm, J.; Weill, G. Persistence length of single-stranded DNA. Macromolecules 1997, 30, 5763–5765. [Google Scholar]
- Kuszewski, J.; Gronenborn, A.M.; Clore, G.M. Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration. J. Am. Chem. Soc. 1999, 121, 2337–2338. [Google Scholar] [CrossRef]
- Manning, G.S. Limiting laws and counterion condensation in polyelectrolyte solutions. 7. Electrophoretic mobility and conductance. J. Phys. Chem. 1981, 85, 1506–1515. [Google Scholar]
- Rechendorff, K.; Witz, G.; Adamcik, J.; Dietler, G. Persistence length and scaling properties of single-stranded DNA adsorbed on modified graphite. J. Chem. Phys. 2009, 131, 095103:1–095103:6. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qian, W.; Doi, K.; Uehara, S.; Morita, K.; Kawano, S. Theoretical Study of the Transpore Velocity Control of Single-Stranded DNA. Int. J. Mol. Sci. 2014, 15, 13817-13832. https://doi.org/10.3390/ijms150813817
Qian W, Doi K, Uehara S, Morita K, Kawano S. Theoretical Study of the Transpore Velocity Control of Single-Stranded DNA. International Journal of Molecular Sciences. 2014; 15(8):13817-13832. https://doi.org/10.3390/ijms150813817
Chicago/Turabian StyleQian, Weixin, Kentaro Doi, Satoshi Uehara, Kaito Morita, and Satoyuki Kawano. 2014. "Theoretical Study of the Transpore Velocity Control of Single-Stranded DNA" International Journal of Molecular Sciences 15, no. 8: 13817-13832. https://doi.org/10.3390/ijms150813817