Rapamycin-Induced Apoptosis in HGF-Stimulated Lens Epithelial Cells by AKT/mTOR, ERK and JAK2/STAT3 Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
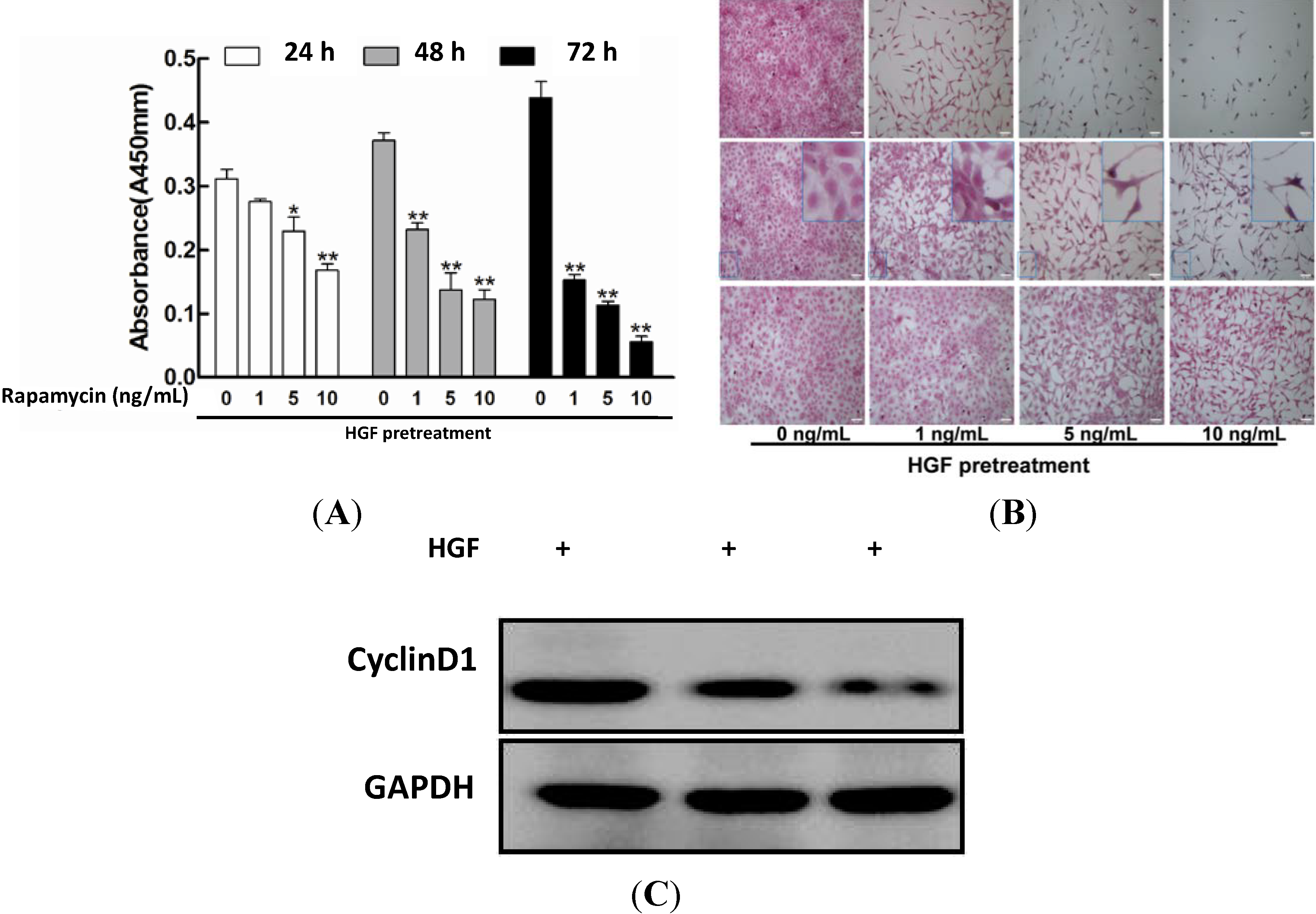
2.1. Impact of Rapamycin on the Proliferation in HGF-Treated Lens Epithelial Cells (LECs)

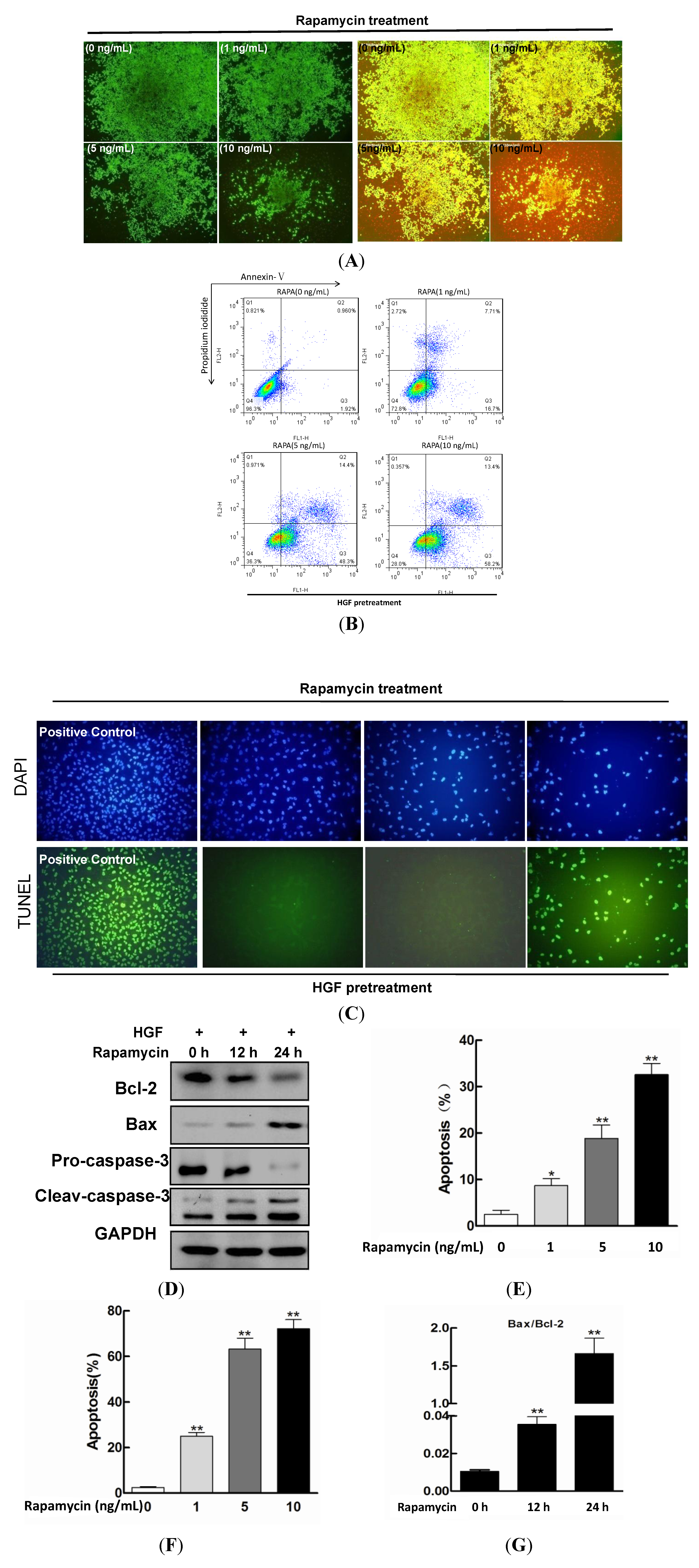
2.2. Effect of Rapamycin on the Apoptosis in HGF-Treated LECs

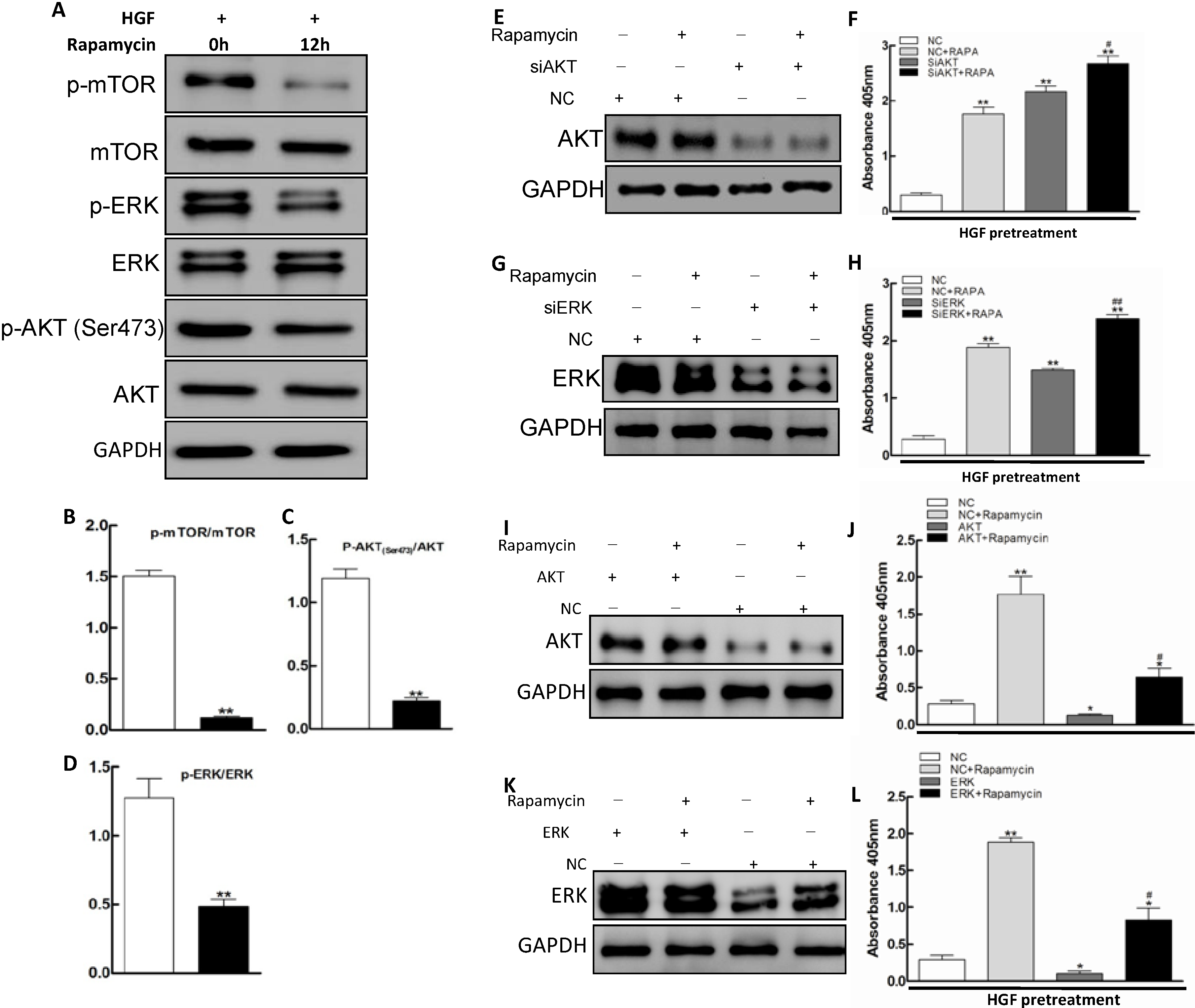
2.3. AKT/mTOR and ERK Pathways Involvement in Apoptosis Promoted by Rapamycin in HGF-Treated LECs

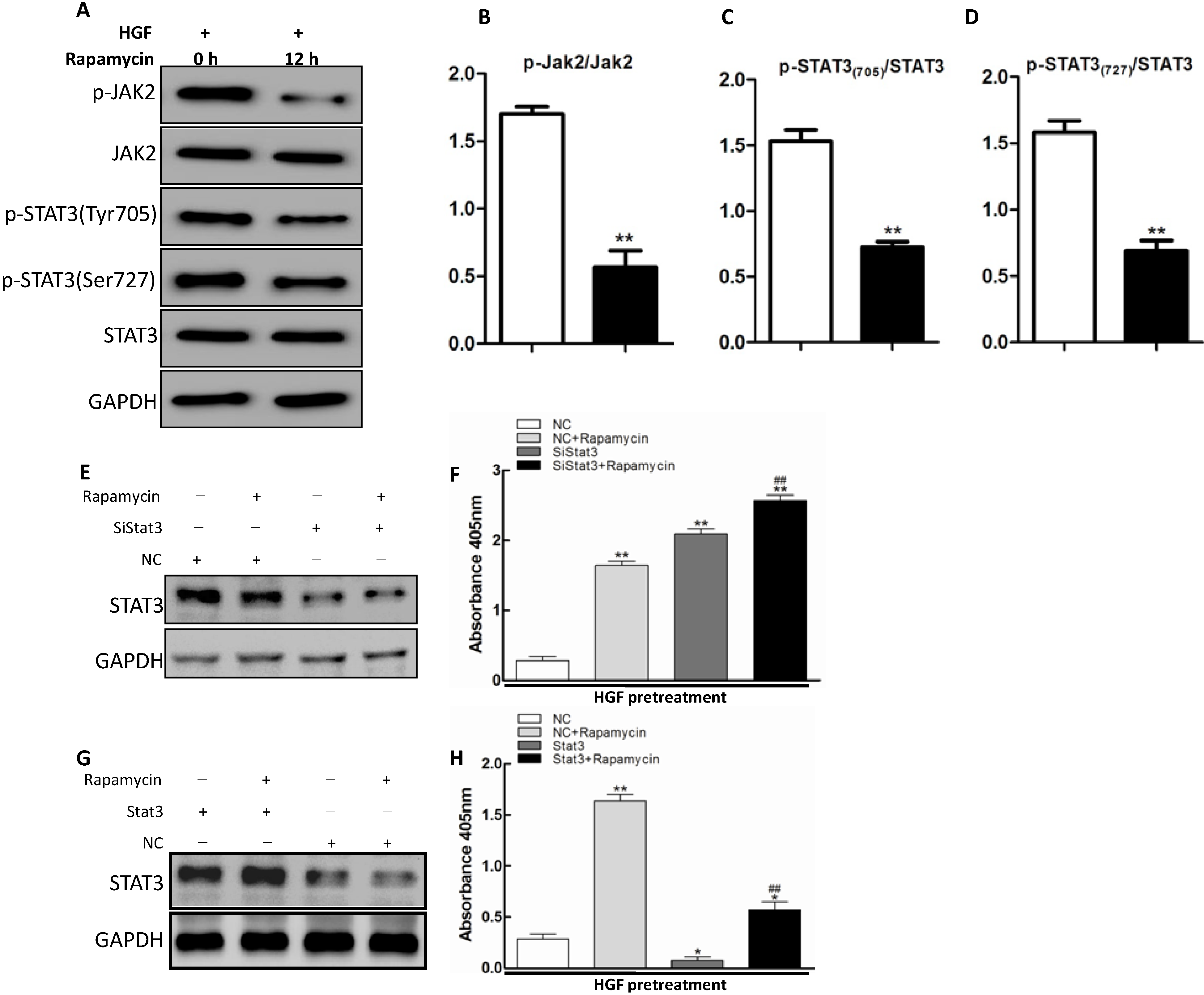
2.4. JAK2/STAT3 Pathway Involvement in Apoptosis Promoted by Rapamycin in HGF-Treated LECs
3. Experimental Section
3.1. Cell Culture, RNA Interference and Transient Transfection
3.2. Proliferation Assay
3.3. Cell Viability Assays

3.4. Apoptosis Assays
3.5. Western Blot Analysis
3.6. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Awasthi, N.; Wagner, B.J. Suppression of human lens epithelial cell proliferation by proteasome inhibition, a potential defense against posterior capsular opacification. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4482–4489. [Google Scholar]
- Apple, D.J.; Solomon, K.D.; Tetz, M.R.; Assia, E.I.; Holland, E.Y.; Legler, U.F.; Tsai, J.C.; Castaneda, V.E.; Hoggatt, J.P.; Kostick, A.M. Posterior capsule opacification. Surv. Ophthalmol. 1992, 37, 73–116. [Google Scholar]
- Choi, J.; Park, S.Y.; Joo, C.K. Hepatocyte growth factor induces proliferation of lens epithelial cells through activation of ERK1/2 and JNK/SAPK. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2696–2704. [Google Scholar]
- Hodge, W.G. Posterior capsule opacification after cataract surgery. Ophthalmology 1998, 105, 943–944. [Google Scholar]
- McDonnell, P.J.; Krause, W.; Glaser, B.M. In vitro inhibition of lens epithelial cell proliferation and migration. Ophthalmic Surg. 1988, 19, 25–30. [Google Scholar]
- Apple, D.J.; Peng, Q.; Visessook, N.; Werner, L.; Pandey, S.K.; Escobar-Gomez, M.; Ram, J.; Auffarth, G.U. Eradication of posterior capsule opacification: documentation of a marked decrease in Nd:YAG laser posterior capsulotomy rates noted in an analysis of 5416 pseudophakic human eyes obtained postmortem. Ophthalmology 2001, 108, 505–518. [Google Scholar]
- Javitt, J.C.; Tielsch, J.M.; Canner, J.K.; Kolb, M.M.; Sommer, A.; Steinberg, E.P. National outcomes of cataract extraction. Increased risk of retinal complications associated with Nd:YAG laser capsulotomy. The Cataract Patient Outcomes Research Team. Ophthalmology 1992, 99, 1487–1497. [Google Scholar]
- Wallentin, N.; Wickstrom, K.; Lundberg, C. Effect of cataract surgery on aqueous TGF-beta and lens epithelial cell proliferation. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1410–1418. [Google Scholar]
- Meacock, W.R.; Spalton, D.J.; Stanford, M.R. Role of cytokines in the pathogenesis of posterior capsule opacification. Br. J. Ophthalmol. 2000, 84, 332–336. [Google Scholar]
- Wormstone, I.M.; Tamiya, S.; Marcantonio, J.M.; Reddan, J.R. Hepatocyte growth factor function and c-Met expression in human lens epithelial cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4216–4222. [Google Scholar]
- Duncan, G.; Wormstone, I.M.; Liu, C.S.; Marcantonio, J.M.; Davies, P.D. Thapsigargin-coated intraocular lenses inhibit human lens cell growth. Nat. Med. 1997, 3, 1026–1028. [Google Scholar]
- Dai, Z.J.; Gao, J.; Ma, X.B.; Kang, H.F.; Wang, B.F.; Lu, W.F.; Lin, S.; Wang, X.J.; Wu, W.Y. Antitumor effects of rapamycin in pancreatic cancer cells by inducing apoptosis and autophagy. Int. J. Mol. Sci. 2012, 14, 273–285. [Google Scholar]
- Morris, R.E. Rapamycin: Antifungal, antitumor, antiproliferative and immunosuppressive macrolide. Elsevier 1992, 6, 39–87. [Google Scholar]
- Liu, H.; Feng, G.; Wu, L.; Fu, S.; Liu, P.; Yang, W.; Zhang, X. The effects of rapamycin on lens epithelial cell proliferation, migration, and matrix formation: an in vitro study. Mol. Vis. 2010, 16, 1646–1653. [Google Scholar]
- Liu, H.; Wu, L.; Fu, S.; Hou, Y.; Liu, P.; Cui, H.; Liu, J.; Xing, L.; Zhang, X. Polylactide-glycoli acid and rapamycin coating intraocular lens prevent posterior capsular opacification in rabbit eyes. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 801–807. [Google Scholar]
- Wang, Z.; Wang, Z. Effects of rapamycin on expression of Bcl-2 and Bax in human lens epithelial cells and cell cycle in rats. J. Huazhong Univ. Sci. Technol. Med. Sci. 2011, 31, 555–559. [Google Scholar]
- Svirshchevskaya, E.V.; Mariotti, J.; Wright, M.H.; Viskova, N.Y.; Telford, W.; Fowler, D.H.; Varticovski, L. Rapamycin delays growth of Wnt-1 tumors in spite of suppression of host immunity. BMC Cancer 2008, 8, 1471–2407. [Google Scholar]
- Wu, Q.; Kiguchi, K.; Kawamoto, T.; Ajiki, T.; Traag, J.; Carbajal, S.; Ruffino, L.; Thames, H.; Wistuba, I.; Thomas, M.; et al. Therapeutic effect of rapamycin on gallbladder cancer in a transgenic mouse model. Cancer Res. 2007, 67, 3794–800. [Google Scholar] [CrossRef]
- Butzal, M.; Logesa, S.; Schweizerb, M.; Fischera, U.; Gehlinga, U.M.; Hossfelda, D.K.; Fiedlera, W. Rapamycin inhibits proliferation and differentiation of huma n endothelial progenitor cells in vitro. Exp. Cell Res. 2004, 300, 65–71. [Google Scholar]
- Morikawa, Y.; Koike, H.; Sekine, Y.; Matsui, H.; Shibata, Y.; Ito, K.; Suzuki, K. Rapamycin enhances docetaxel-induced cytotoxicity in a androgen-independent prostate cancer xenograft model by survivin downregulation. Biochem. Biophys. Res. Commun. 2012, 419, 584–589. [Google Scholar]
- Dai, Z.J.; Gao, J.; Kang, H.F.; Ma, Y.G.; Ma, X.B.; Lu, W.F.; Lin, S.; Ma, H.B.; Wang, X.J.; Wu, W.Y. Targeted inhibition of mammalian target of rapamycin (mTOR) enhances radiosensitivity in pancreatic carcinoma cells. Drug Des. Dev. Ther. 2013, 7, 149–159. [Google Scholar]
- Di Paolo, S.; Teutonico, A.; Ranieri, E.; Gesualdo, L.; Schena, P.F. Monitoring antitumor efficacy of rapamycin in Kaposi sarcoma. Am. J. Kidney Dis. 2007, 49, 462–470. [Google Scholar]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef]
- Meng, Q.; Guo, H.; Xiao, L.; Cui, Y.; Guo, R.; Xiao, D.; Huang, Y. mTOR regulates TGF-β2-induced epithelial-mesenchymal transition in cultured human lens epithelial cells. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 2363–2370. [Google Scholar]
- Oltvai, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Marzo, I.; Brenner, C.; Zamzami, N.; Jurgensmeier, J.M.; Susin, S.A.; Vieira, H.L.; Prevost, M.C.; Xie, Z.; Matsuyama, S.; Reed, J.C.; et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 1998, 281, 2027–2031. [Google Scholar] [CrossRef]
- Fang, Z.; Tang, Y.; Fang, J.; Zhou, Z.; Xing, Z.; Guo, Z.; Guo, X.; Wang, W.; Jiao, W.; Xu, Z.; et al. Simvastatin inhibits renal cancer cell growth and metastasis via AKT/mTOR, ERK and JAK2/STAT3 pathway. PLoS One 2013, 8, e62823. [Google Scholar]
- Malecaze, F.; Decha, A.; Serre, B.; Penary, M.; Duboue, M.; Berg, D.; Levade, T.; Lubsen, N.H.; Kremer, E.J.; Couderc, B. Prevention of posterior capsule opacification by the induction of therapeutic apoptosis of residual lens cells. Gene Ther. 2006, 13, 440–448. [Google Scholar]
- Wu, X.H.; Lu, Y.; Fang, Y.W.; Jiang, Y.X. The polyamidoamine-mediated inhibition of bcl-2 by small hairpin RNA to induce apoptosis in human lens epithelial cells. Mol. Vis. 2012, 18, 74–80. [Google Scholar]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar]
- Vigna, E.; Naldini, L.; Tamagnone, L.; Longati, P.; Bardelli, A.; Maina, F.; Ponzetto, C.; Comoglio, P. Hepatocyte growth factor and its receptor, the tyrosine kinase encoded by the c-MET proto-oncogene. Cell. Mol. Biol. 1994, 40, 597–604. [Google Scholar]
- Weidner, K.M.; Hartmann, G.; Sachs, M.; Birchmeier, W. Properties and Functions of Scatter Factor Hepatocyte Growth Factor and Its Receptor c-Met. Am. J. Respir. Cell Mol. Biol. 1993, 8, 229–237. [Google Scholar]
- Zarnegar, R.; Michalopoulos, G.K. The many faces of hepatocyte growth factor: From hepatopoiesis to hematopoiesis. J. Cell. Biol. 1995, 129, 1177–1180. [Google Scholar]
- Reed, J.C.; Pellecchia, M. Apoptosis-based therapies for hematologic malignancies. Blood 2005, 106, 408–418. [Google Scholar]
- Yang, J.C.; Cortopassi, G.A. Induction of the mitochondrial permeability transition causes release of the apoptogenic factor cytochrome c. Free Radic. Biol. Med. 1998, 24, 624–631. [Google Scholar]
- Lei, F.R.; Li, X.Q.; Liu, H.; Zhu, R.D.; Meng, Q.Y.; Rong, J.J. Rapamycin and 3-methyladenine regulate apoptosis and autophagy in bone-derived endothelial progenitor cells. Chin. Med. J. 2012, 125, 4076–4082. [Google Scholar]
- Zhang, Y.; Zhang, J.W.; Lv, G.; Xie, S.; Wang, G. Effects of STAT3 gene silencing and rapamycin on apoptosis in hepatocarcinoma cells. Int. J. Med. Sci. 2012, 9, 216–224. [Google Scholar]
- Cai, N.; Dai, S.D.; Liu, N.N.; Liu, L.M.; Zhao, N.; Chen, L. PI3K/AKT/mTOR signaling pathway inhibitors in proliferation of retinal pigment epithelial cells. Int. J. Ophthalmol. 2012, 5, 675–680. [Google Scholar]
- Pelaia, G.; Gallelli, L.; Renda, T.; Fratto, D.; Falcone, D.; Caraglia, M.; Busceti, M.T.; Terracciano, R.; Vatrella, A.; Maselli, R.; et al. Effects of statins and farnesyl transferase inhibitors on ERK phosphorylation, apoptosis and cell viability in non-small lung cancer cells. Cell Prolif. 2012, 45, 557–565. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Kimlinger, T.; Haug, J.; Painuly, U.; Wellik, L.; Halling, T.; Rajkumar, S.V.; Kumar, S. Anti-myeloma activity of AKT inhibition is linked to the activation status of PI3K/AKT and MEK/ERK pathway. PLoS One 2012, 7, e50005. [Google Scholar]
- Hsieh, F.C.; Cheng, G.; Lin, J. Evaluation of potential STAT3-regulated genes in human breast cancer. Biochem. Biophys. Res. Commun. 2005, 335, 292–299. [Google Scholar]
- Ebong, S.; Yu, C.R.; Carper, D.A.; Chepelinsky, A.B.; Egwuagu, C.E. Activation of STAT signaling pathways and induction of suppressors of cytokine signaling (SOCS) proteins in mammalian lens by growth factors. Investig. Ophthalmol. Vis. Sci. 2004, 45, 872–878. [Google Scholar]
- Das, A.; Salloum, F.N.; Durrant, D.; Ockaili, R.; Kukreja, R.C. Rapamycin protects against myocardial ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J. Mol. Cell. Cardiol. 2012, 53, 858–869. [Google Scholar] [CrossRef]
- Fuglesteg, B.N.; Tiron, C.; Jonassen, A.K.; Mjos, O.D.; Ytrehus, K. Pretreatment with insulin before ischaemia reduces infarct size in Langendorff-perfused rat hearts. Acta Physiol. 2009, 195, 273–282. [Google Scholar]
- Hausenloy, D.J.; Mocanu, M.M.; Yellon, D.M. Cross-talk between the survival kinases during early reperfusion: Its contribution to ischemic preconditioning. Cardiovasc. Res. 2004, 63, 305–312. [Google Scholar]
- Boengler, K.; Hilfiker-Kleiner, D.; Drexler, H.; Heusch, G.; Schulz, R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol. Ther. 2008, 120, 172–185. [Google Scholar]
- Bolli, R.; Stein, A.B.; Guo, Y.; Wang, O.L.; Rokosh, G.; Dawn, B.; Molkentin, J.D.; Sanganalmath, S.K.; Zhu, Y.; Xuan, Y.T. A murine model of inducible, cardiac-specific deletion of STAT3: Its use to determine the role of STAT3 in the upregulation of cardioprotective proteins by ischemic preconditioning. J. Mol. Cell. Cardiol. 2011, 50, 589–597. [Google Scholar]
- Khan, S.; Salloum, F.; Das, A.; Xi, L.; Vetrovec, G.W.; Kukreja, R.C. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 41, 256–264. [Google Scholar]
- Jonassen, A.K.; Sack, M.N.; Mjos, O.D.; Yellon, D.M. Myocardial protection by insulin at reperfusion requires early administration and is mediated via AKT and p70s6 kinase cell-survival signaling. Circ. Res. 2001, 89, 1191–1198. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tian, F.; Dong, L.; Zhou, Y.; Shao, Y.; Li, W.; Zhang, H.; Wang, F. Rapamycin-Induced Apoptosis in HGF-Stimulated Lens Epithelial Cells by AKT/mTOR, ERK and JAK2/STAT3 Pathways. Int. J. Mol. Sci. 2014, 15, 13833-13848. https://doi.org/10.3390/ijms150813833
Tian F, Dong L, Zhou Y, Shao Y, Li W, Zhang H, Wang F. Rapamycin-Induced Apoptosis in HGF-Stimulated Lens Epithelial Cells by AKT/mTOR, ERK and JAK2/STAT3 Pathways. International Journal of Molecular Sciences. 2014; 15(8):13833-13848. https://doi.org/10.3390/ijms150813833
Chicago/Turabian StyleTian, Fang, Lijie Dong, Yu Zhou, Yan Shao, Wenbo Li, Hong Zhang, and Fei Wang. 2014. "Rapamycin-Induced Apoptosis in HGF-Stimulated Lens Epithelial Cells by AKT/mTOR, ERK and JAK2/STAT3 Pathways" International Journal of Molecular Sciences 15, no. 8: 13833-13848. https://doi.org/10.3390/ijms150813833