Heterosis in Early Maize Ear Inflorescence Development: A Genome-Wide Transcription Analysis for Two Maize Inbred Lines and Their Hybrid

Abstract
:1. Introduction
2. Results
2.1. Statistics and Analysis of Library Sequencing

{kind=link}
{kind=link}
{kind=link}
| Class | Summary | F1 | B73 | Mo17 |
|---|---|---|---|---|
| Raw Data | Total | 4,200,000 | 4,200,000 | 4,200,000 |
| Distinct Tags | 272,402 | 254,882 | 252,386 | |
| Clean Tags | Total number | 4,176,622 | 4,176,825 | 4,176,752 |
| Distinct Tags number | 259,282 | 242,184 | 239,963 | |
| All Tags Mapping to Genome | Total number | 2,766,685 | 2,979,615 | 3,127,520 |
| Total % of clean tags | 66.24% | 71.34% | 74.88% | |
| Distinct Tags number | 134,449 | 134,378 | 141,476 | |
| Distinct Tags % of clean tags | 51.85% | 55.49% | 58.96% | |
| Unambiguous Tags Mapping to Genome | Total number | 2,452,293 | 2,637,419 | 2,804,389 |
| Total % of clean tags | 58.71% | 63.14% | 67.14% | |
| Distinct Tags number | 119,316 | 118,719 | 125,467 | |
| Distinct Tags % of clean tags | 46.02% | 49.02% | 52.29% | |
| All Tags Mapping to Genes | Number | 24,629 | 24,078 | 24,198 |
| % of ref. genes | 75.69% | 74% | 74.36% | |
| Unambiguous Tags Mapping to Genes | Number | 21,372 | 20,784 | 20,938 |
| % of ref. genes | 65.68% | 63.87% | 64.35% | |
| Unknown Tags | Total number | 894,503 | 721,786 | 561,476 |
| Total % of clean tags | 21.42% | 17.28% | 13.44% | |
| Distinct Tags number | 85,152 | 70,700 | 61,016 | |
| Distinct Tags % of clean tags | 32.84% | 29.19% | 25.43% |
2.2. Mapping Tags to the Maize Reference Genome
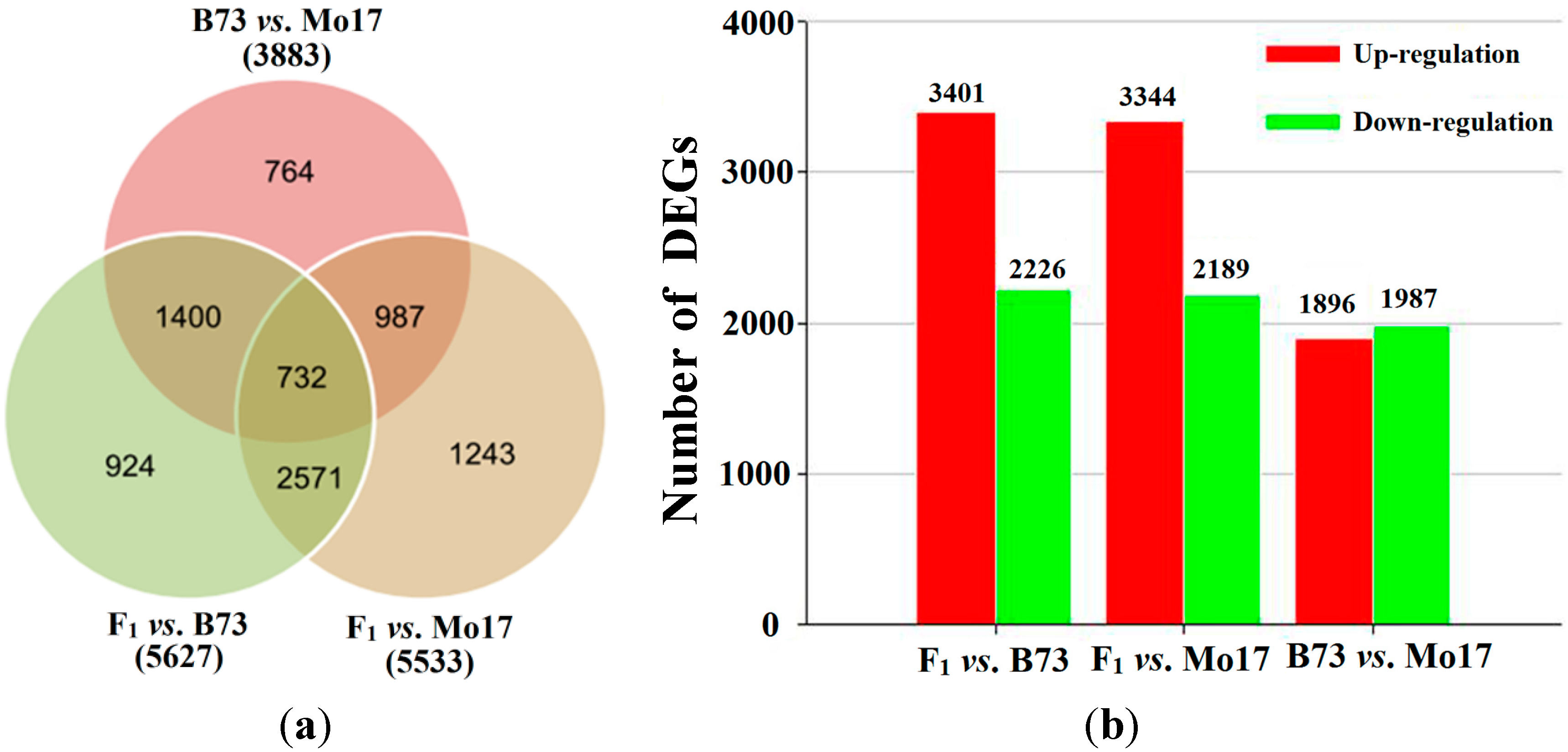
2.3. Different Gene Expression Analysis


| Hybrid Cross | B73 × Mo17 |
|---|---|
| Total | 8621 |
| Additivity | 767 |
| Non Additivity | 7854 |
| High-Parent Dominance | 1984 |
| Low-Parent Dominance | 2559 |
| Over Dominance | 1085 |
| Under Dominance | 1963 |
| Other | 263 |

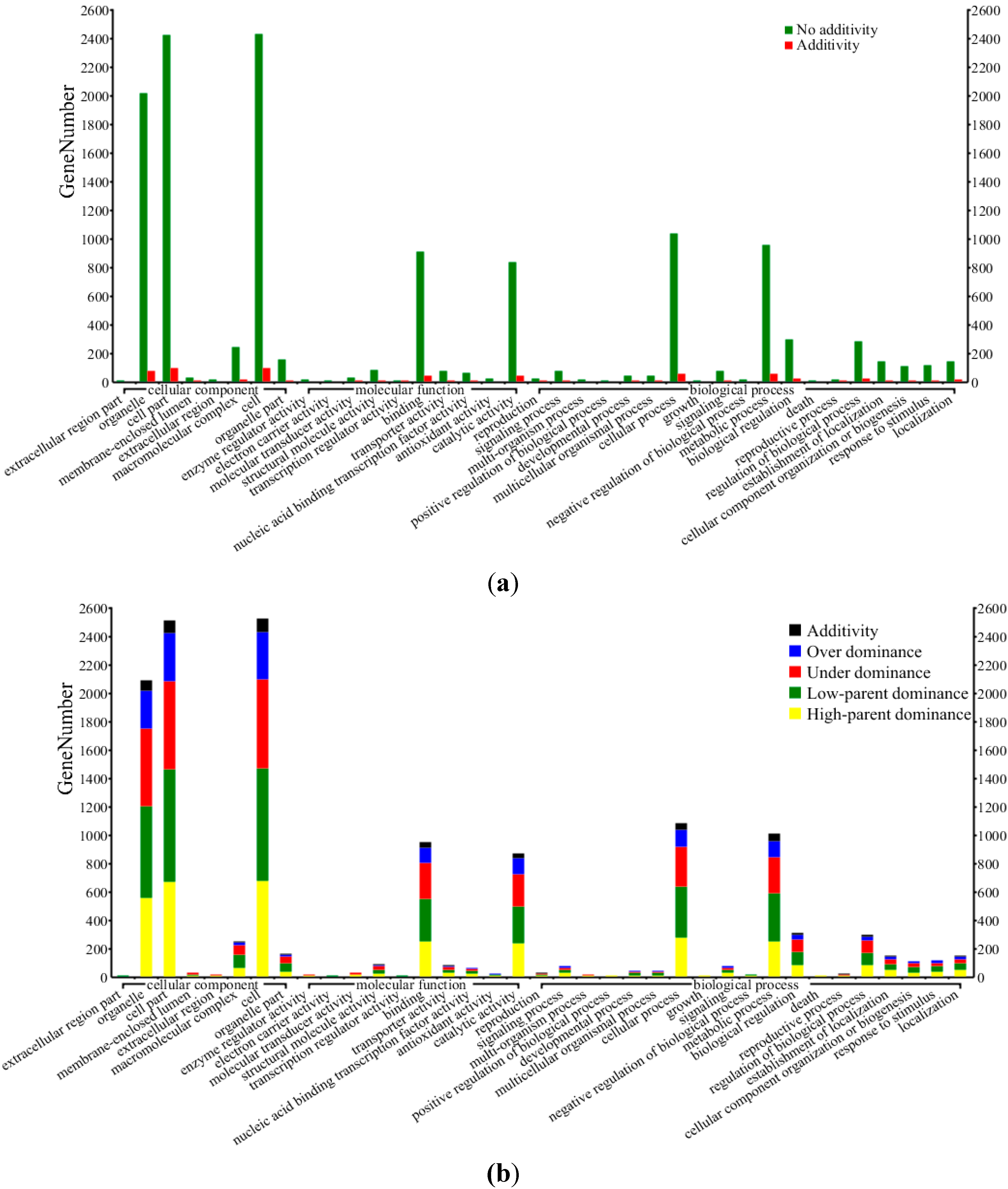
2.4. Functional Enrichment Analysis for Differentially Expressed Genes Using Gene Ontology (GO)
| Functional Categories | Additivity | Non-Additivity | HPD | LPD | UDO | ODO | Other |
|---|---|---|---|---|---|---|---|
| Cellular Component | |||||||
| Extracellular Region Part | 1 | 3 | 0 | 2 | 0 | 1 | 0 |
| Organelle * | 181 | 2077 | 544 | 650 | 542 | 271 | 70 |
| Cell Part | 221 | 2499 | 655 | 793 | 624 | 339 | 88 |
| Membrane-Enclosed Lumen | 1 | 24 | 3 | 9 | 10 | 1 | 1 |
| Extracellular Region | 1 | 13 | 1 | 5 | 2 | 5 | 0 |
| Macromolecular Complex * | 16 | 245 | 58 | 89 | 71 | 16 | 11 |
| Cell | 221 | 2510 | 660 | 794 | 628 | 340 | 88 |
| Organelle Part | 16 | 155 | 31 | 57 | 47 | 14 | 6 |
| Molecular Function | |||||||
| Enzyme Regulator Activity | 3 | 12 | 3 | 2 | 3 | 4 | 0 |
| Electron Carrier Activity | 0 | 3 | 0 | 1 | 2 | 0 | 0 |
| Molecular Transducer Activity | 3 | 24 | 7 | 5 | 9 | 2 | 1 |
| Structural Molecule Activity | 3 | 80 | 17 | 27 | 28 | 4 | 4 |
| Transcription Regulator Activity | 2 | 6 | 0 | 1 | 2 | 2 | 1 |
| Binding | 75 | 940 | 241 | 297 | 258 | 108 | 36 |
| Transporter Activity | 9 | 76 | 25 | 19 | 20 | 9 | 3 |
| Nucleic Acid Binding Transcription Factor Activity | 2 | 58 | 18 | 19 | 15 | 2 | 4 |
| Antioxidant Activity | 0 | 17 | 5 | 3 | 3 | 6 | 0 |
| Catalytic Activity | 67 | 865 | 229 | 263 | 226 | 111 | 36 |
| Biological Process | |||||||
| Reproduction | 0 | 21 | 3 | 8 | 5 | 3 | 2 |
| Signaling Process | 5 | 70 | 24 | 20 | 16 | 7 | 3 |
| Multi-Organism Process | 0 | 8 | 4 | 2 | 1 | 1 | 0 |
| Positive Regulation of Biological Process | 0 | 3 | 2 | 1 | 0 | 0 | 0 |
| Developmental Process | 2 | 38 | 2 | 21 | 9 | 4 | 2 |
| Multicellular Organismal Process | 2 | 39 | 4 | 20 | 9 | 4 | 2 |
| Cellular Process | 71 | 1079 | 269 | 357 | 281 | 125 | 47 |
| Growth | 0 | 5 | 1 | 2 | 1 | 1 | 0 |
| Signaling | 5 | 70 | 24 | 20 | 16 | 7 | 3 |
| Negative Regulation of Biological Process | 2 | 12 | 2 | 6 | 3 | 1 | 0 |
| Metabolic Process | 70 | 999 | 242 | 338 | 253 | 118 | 48 |
| Biological Regulation | 18 | 303 | 77 | 94 | 88 | 28 | 16 |
| Death | 2 | 6 | 2 | 1 | 2 | 1 | 0 |
| Reproductive Process | 0 | 15 | 1 | 5 | 5 | 2 | 2 |
| Regulation of Biological Process | 17 | 289 | 76 | 88 | 84 | 26 | 15 |
| Establishment of Localization | 15 | 143 | 41 | 44 | 33 | 19 | 6 |
| Cellular Component Organization or Biogenesis | 10 | 106 | 24 | 38 | 29 | 10 | 5 |
| Response to Stimulus | 4 | 112 | 33 | 37 | 22 | 19 | 1 |
| Localization | 16 | 145 | 41 | 45 | 33 | 19 | 7 |
| GO Term | Definition | FDR Corrected p-Value a |
|---|---|---|
| Cellular Component | ||
| GO:0043228 | Non-membrane-bounded organelle | 6.40 × 10−4 |
| GO:0043232 | Intracellular non-membrane-bounded organelle | 6.40 × 10−4 |
| GO:0005634 | Nucleus | 2.56 × 10−3 |
| GO:0005840 | Ribosome | 3.34 × 10−2 |
| GO:0032991 | Macromolecular complex | 4.27 × 10−2 |
| Biological Process | ||
| GO:0044267 | Cellular protein metabolic process | 4.22 × 10−3 |
| GO:0044260 | Cellular macromolecule metabolic process | 8.84 × 10−3 |
| GO:0019538 | Protein metabolic process | 3.30 × 10−2 |
| GO:0009987 | Cellular process | 3.37 × 10−2 |
| GO:0043170 | Macromolecule metabolic process | 4.59 × 10−2 |
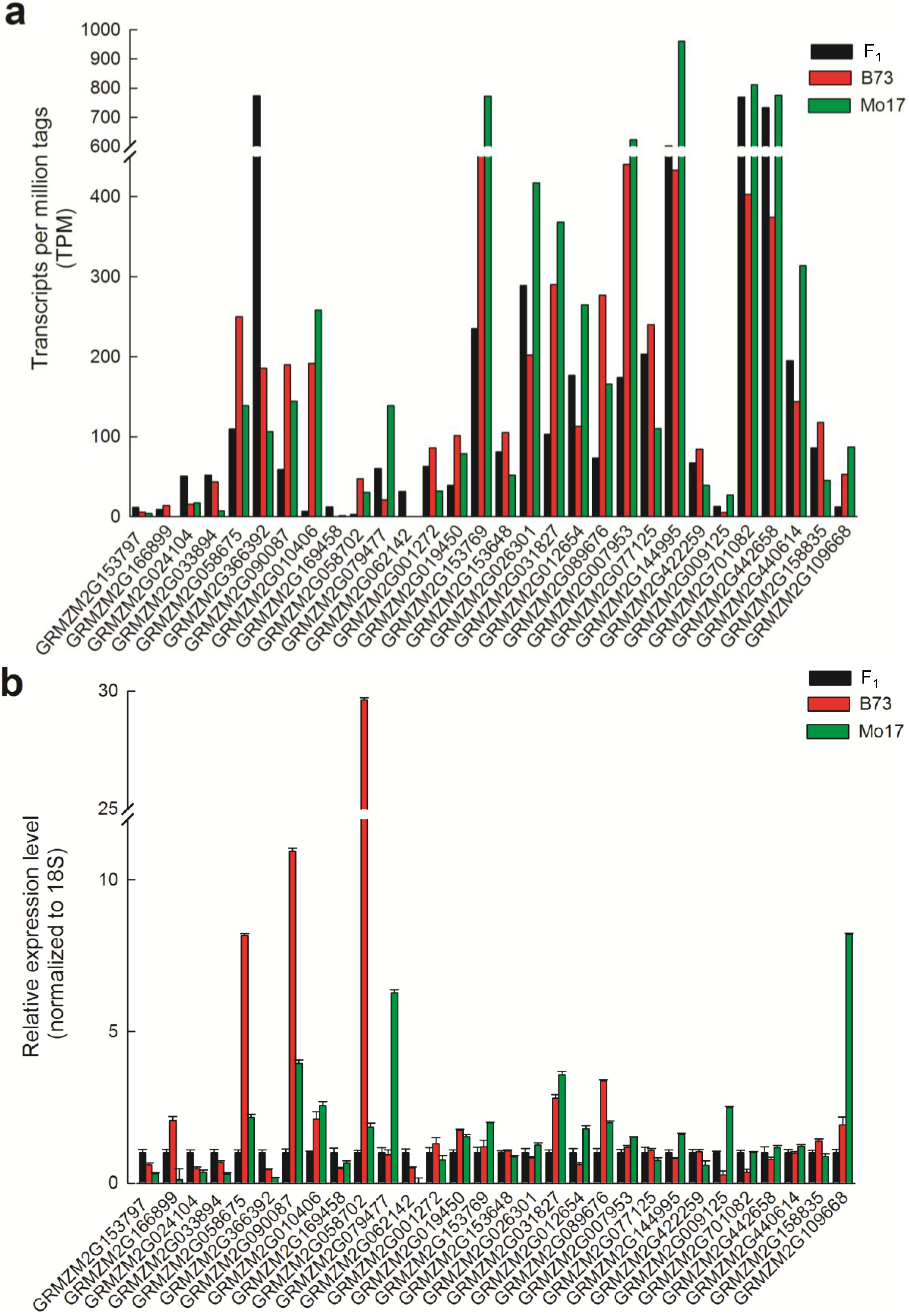
2.5. The Expression Patterns of Biological Macromolecular Synthesis-Related Genes in the B73 × Mo17 Hybrid
| Pathway a | Different Genes with Pathway Annotation (4312) | p -Value b | q -Value c | Pathway ID | |||
|---|---|---|---|---|---|---|---|
| F1 vs. B73 | F1 vs. Mo17 | Overlap | Total (%) | ||||
| Ribosome ** | 148 | 129 | 89 | 188 (4.68%) | 2.47 × 10−5 | 1.58 × 10−3 | ko03010 |
| Arginine and Proline Metabolism ** | 37 | 34 | 25 | 46 (1.11%) | 2.59 × 10−5 | 1.58 × 10−3 | ko00330 |
| Spliceosome * | 145 | 136 | 87 | 194 (4.82%) | 4.01 × 10−4 | 1.63 × 10−2 | ko03040 |
| Pyruvate Metabolism * | 32 | 41 | 25 | 49 (1.69%) | 6.28 × 10−4 | 3.80 × 10−2 | ko00620 |
| Proteasome | 33 | 25 | 19 | 39 (0.90%) | 2.00 × 10−3 | 6.09 × 10−2 | ko03050 |
| Pentose Phosphate Pathway | 16 | 24 | 13 | 27 (0.63%) | 1.66 × 10−3 | 6.70 × 10−2 | ko00030 |
| Purine Metabolism | 64 | 72 | 39 | 97 (2.25%) | 9.34 × 10−4 | 7.50 × 10−2 | ko00230 |
| RNA Degradation | 43 | 49 | 31 | 61 (1.41%) | 4.38 × 10−3 | 8.84 × 10−2 | ko03018 |
| Porphyrin and Chlorophyll Metabolism | 21 | 16 | 11 | 26 (0.60%) | 3.82 × 10−3 | 9.32 × 10−2 | ko00860 |
| Glycolysis/Gluconeogenesis | 49 | 57 | 31 | 75 (1.74%) | 8.38 × 10−3 | 1.04 × 10−1 | ko00010 |
2.6. Resolving Transcription Factors (TFs) among Differentially Expressed Genes
| Maize Gene ID | Annotation a | TF Family | Significant Pattern b | Expression Model c | Regulated in ra3 Mutants d |
|---|---|---|---|---|---|
| GRMZM2G106673 | B3-domain TF | B3 | Mo17 < F1 ≈ B73 | HPD | Up |
| GRMZM2G177046 | Ocs element-binding factor 1 | bZIP | Mo17 < F1 ≈ B73 | HPD | Up |
| GRMZM2G102514 | BES1/BZR1 protein | BES | Mo17 < F1 ≈ B73 | HPD | Down |
| GRMZM2G172657 | Uncharacterized | GRAS | Mo17 < F1 ≈ B73 | HPD | Down |
| GRMZM2G115357 | IAA24 | AUX/IAA | Mo17 < F1 ≈ B73 | HPD | Up |
| GRMZM2G181376 | Uncharacterized | – | B73 < F1 ≈ Mo17 | HPD | Down |
| GRMZM2G173534 | Inducer of CBF expression 2 | – | B73 < F1 ≈ Mo17 | HPD | Up |
| GRMZM2G173124 | Zinc finger | Znf-C3H1 | B73 ≈ F1 < Mo17 | LPD | Up |
| GRMZM2G138886 | Cyclin B2 | Cyclin | B73 ≈ F1 < Mo17 | LPD | Up |
| GRMZM2G020054 | Uncharacterized | AP2/ERF | Mo17 ≈ F1 < B73 | LPD | Up |
| GRMZM2G055243 | KNOX class 2 protein | KNOX | Mo17 ≈ F1 < B73 | LPD | Up |
| GRMZM2G089995 | Ethylene responsive | AP2/ERF | Mo17 ≈ F1 < B73 | LPD | Up |
| GRMZM2G079825 | Pathogenesis-related | AP2/ERF | Mo17 ≈ F1 < B73 | LPD | NS |
| GRMZM2G310368 | Ethylene responsive | AP2/ERF | Mo17 ≈ F1 < B73 | LPD | Up |
| GRMZM2G132185 | Pathogenesis-related | AP2/ERF | Mo17 ≈ F1 < B73 | LPD | NS |
| GRMZM2G149940 | B3 DNA binding domain | B3 | Mo17 ≈ F1 < B73 | LPD | Up |
| GRMZM2G003927 | Ramosa1 | Znf-C2H2 | B73 ≈ F1 < Mo17 | LPD | Up |
| GRMZM2G447406 | Progesterone receptor | – | B73 ≈ F1 < Mo17 | LPD | Down |
| GRMZM2G102218 | YABBY protein | YABBY | Mo17 ≈ F1 < B73 | LPD | Up |
| GRMZM2G136769 | Ubiquitin-associated | Ubiquitin | Mo17 ≈ F1 < B73 | LPD | Up |
| GRMZM2G165972 | Heat shock factor (HSF)-type | HSF | Mo17 ≈ F1 < B73 | LPD | Down |
| GRMZM2G140474 | Tyrosine protein kinase | – | Mo17 ≈ F1 < B73 | LPD | Up |
| GRMZM2G422205 | Zinc finger | Znf-C3H1 | Mo17 ≈ F1 < B73 | LPD | |
| GRMZM2G171468 | Uncharacterized | MYB | Mo17 ≈ F1 < B73 | LPD | Down |
| AC206951.3_FG017 | Ethylene-responsive element binding protein 2 | ERF | Mo17 < B73 < F1 | ODO | Up |
| GRMZM2G081012 | Transcription initiation factor IID, 18 kD subunit family protein | TFIID-18 | B73 ≈ Mo17 < F1 | ODO | Up |
| GRMZM2G014653 | NAC protein 48 | NAC | B73 ≈ Mo17 < F1 | ODO | Up |
| GRMZM2G127379 | NAM containing | NAC | B73 ≈ Mo17 < F1 | ODO | Up |
| GRMZM2G061487 | DRE binding factor 1 | AP2/ERF | F1 < Mo17 < B73 | UDO | Up |
| GRMZM2G431157 | Zinc finger, C2H2-type | Znf_C2H2-like | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G307119 | Branched silkless1 | AP2/ERF | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G144275 | bHLH transcription factor | HLH | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G132367 | HDZipI-1 | HD-Zip | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G105266 | Pathogenesis-related | AP2/ERF | F1 < B73 ≈ Mo17 | UDO | NS |
| GRMZM2G381395 | DNA-directed RNA polymerase | – | F1 < B73 ≈ Mo17 | UDO | Down |
| GRMZM2G453424 | Uncharacterized | HRDC_like | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G118113 | DNA-directed RNA polymerase II 8.2 kDa polypeptide | EF | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G017606 | SHI | SHI | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G039889 | Cold acclimation protein | – | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G088309 | Drooping leaf | YABBY | F1 < B73 ≈ Mo17 | UDO | Up |
| GRMZM2G078077 | TCP domain protein | TCP | Mo 17 < F1 < B73 | Other | Up |
| GRMZM2G404426 | Zinc finger | Znf-PHD | B73 < F1 < Mo17 | Other | Up |
2.7. A Significant Number of Genes Were Expressed in Only One Inbred Line or Absent in both Inbred Lines
| Class | No. of Genes Present in B73, Absent in Mo17 | % of Genes | No. of Genes Present in Mo17, Absent in B73 | % of Genes | No. of Genes Absent in both B73 and Mo17 | % of Genes |
|---|---|---|---|---|---|---|
| No. of genes present in their hybrid | 942 | 16.6 | 1117 | 19.7 | 1065 | 18.8 |
| No. of genes absent in their hybrid | 1,682 | 29.7 | 854 | 15.1 | 0 | 0 |
| Total | 2,624 | 46.4 | 1971 | 34.8 | 1065 | 18.8 |
2.8. Analysis of Presence/Absence Variation (PAVs) Genes Action in Maize Hybrids
3. Discussion
4. Experimental Section
4.1. Plant Growth and RNA Isolation
4.2. Digital Gene Expression Library Preparation and Sequencing
4.3. Quantitative RT-PCR and Gene Expression Analysis
4.4. Analysis and Mapping of Digital Gene Expression Tags
4.5. Identification of Differentially Expressed Genes
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shull, G.H. The composition of a field of maize. J. Hered. 1908, 4, 296–301. [Google Scholar] [CrossRef]
- Bruce, A.B. The Mendelian theory of heredity and the augmentation of vigor. Science 1910, 32, 627. [Google Scholar]
- Jones, D.F. Dominance of linked factors as a means of accounting for heterosis. Genetics 1917, 6, 466–479. [Google Scholar]
- Davenport, C.B. Degeneration, albinism and inbreeding. Science 1908, 28, 454. [Google Scholar]
- East, E.M. Heterosis. Genetics 1936, 21, 375–397. [Google Scholar]
- Birchler, J.A.; Auger, D.L.; Riddle, N.C. In search of the molecular basis of heterosis. Plant Cell 2003, 15, 2236–2239. [Google Scholar] [CrossRef]
- Yu, S.B.; Li, J.X.; Xu, C.G.; Tan, Y.F.; Gao, Y.J.; Li, X.H.; Zhang, Q.F.; Saghai Maroof, M.A. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 1997, 99, 9226–9231. [Google Scholar]
- Li, Z.K.; Luo, L.J.; Mei, H.W.; Wang, D.L.; Shu, Q.Y.; Tabien, R.; Zhong, D.B.; Ying, C.S.; Stansel, J.W.; Khush, G.S.; et al. Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. I. Biomass and grain yield. Genetics 2001, 158, 1737–1753. [Google Scholar]
- Xiao, J.; Li, J.; Yuan, L.; Tanksley, S.D. Dominance is the major genetic basis of heterosis in rice as revealed by QTL analysis using molecular markers. Genetics 1995, 140, 745–754. [Google Scholar]
- Stuber, C.W.; Lincoln, S.E.; Wolff, D.W.; Helentjaris, T.; Lander, E.S. Identification of genetic factors contributing to heterosis in a hybrid from two elite maize inbred lines using molecular markers. Genetics 1992, 132, 823–839. [Google Scholar]
- Auger, D.L.; Gray, A.D.; Ream, T.S.; Kato, A.; Coe, E.H., Jr.; Birchler, J.A. Nonadditive gene expression in diploid and triploid hybrids of maize. Genetics 2005, 169, 389–397. [Google Scholar]
- Hoecker, N.; Keller, B.; Muthreich, N.; Chollet, D.; Descombes, P.; Piepho, H.P.; Hochholdinger, F. Comparison of maize (Zea mays L.) F1-hybrid and parental inbred line primary root transcriptomes suggests organ-specific patterns of nonadditive gene expression and conserved expression trends. Genetics 2008, 179, 1275–1283. [Google Scholar]
- Song, R.; Messing, J. Gene expression of a gene family in maize based on noncollinear haplotypes. Proc. Natl. Acad. Sci. USA 2003, 100, 9055–9060. [Google Scholar]
- Stupar, R.M.; Springer, N.M. cis-Transcriptional variation in maize inbred lines B73 and Mo17 leads to additive expression patterns in the F1 hybrid. Genetics 2006, 173, 2199–2210. [Google Scholar] [CrossRef]
- Vuylsteke, M.; van Eeuwijk, F.; van Hummelen, P.; Kuiper, M.; Zabeau, M. Genetic analysis of variation in gene expression in Arabidopsis thaliana. Genetics 2005, 171, 1267–1275. [Google Scholar]
- Zhang, H.Y.; He, H.; Chen, L.B.; Li, L.; Liang, M.Z.; Wang, X.F.; Liu, X.G.; He, G.M.; Chen, R.S.; Ma, L.G.; et al. A genome-wide transcription analysis reveals a close correlation of promoter INDEL polymorphism and heterotic gene expression in rice hybrids. Mol. Plant 2008, 1, 720–731. [Google Scholar] [CrossRef]
- Guo, M.; Yang, S.; Rupe, M.; Hu, B.; Bickel, D.; Arthur, L.; Smith, O. Genome-wide allele-specific expression analysis using Massively Parallel Signature Sequencing (MPSS™) Reveals cis- and trans-effects on gene expression in maize hybrid meristem tissue. Plant Mol. Biol. 2008, 66, 551–563. [Google Scholar]
- Chen, X.; Li, M.; Shi, J.; Fu, D.; Qian, W.; Zou, J.; Zhang, C.; Meng, J. Gene expression profiles associated with intersubgenomic heterosis in Brassica napus. Theor. Appl. Genet. 2008, 117, 1031–1040. [Google Scholar] [CrossRef]
- Zhuang, Y.; Adams, K.L. Extensive allelic variation in gene expression in populus F1 hybrids. Genetics 2007, 177, 1987–1996. [Google Scholar] [CrossRef]
- Jahnke, S.; Sarholz, B.; Thiemann, A.; Kühr, V.; Gutiérrez-Marcos, J.; Geiger, H.; Piepho, H.-P.; Scholten, S. Heterosis in early seed development: A comparative study of F1 embryo and endosperm tissues 6 days after fertilization. Theor. Appl. Genet. 2010, 120, 389–400. [Google Scholar] [CrossRef]
- Pea, G.; Ferron, S.; Gianfranceschi, L.; Krajewski, P.; Enrico Pe, M. Gene expression non-additivity in immature ears of a heterotic F1 maize hybrid. Plant Sci. 2008, 174, 17–24. [Google Scholar]
- Riddle, N.; Jiang, H.; An, L.; Doerge, R.; Birchler, J. Gene expression analysis at the intersection of ploidy and hybridity in maize. Theor. Appl. Genet. 2010, 120, 341–353. [Google Scholar] [CrossRef]
- Guo, M.; Rupe, M.A.; Yang, X.; Crasta, O.; Zinselmeier, C.; Smith, O.S.; Bowen, B. Genome-wide transcript analysis of maize hybrids: Allelic additive gene expression and yield heterosis. Theor. Appl. Genet. 2006, 113, 831–845. [Google Scholar] [CrossRef]
- Paschold, A.; Marcon, C.; Hoecker, N.; Hochholdinger, F. Molecular dissection of heterosis manifestation during early maize root development. Theor. Appl. Genet. 2010, 120, 383–388. [Google Scholar] [CrossRef]
- Meyer, S.; Pospisil, H.; Scholten, S. Heterosis associated gene expression in maize embryos 6 days after fertilization exhibits additive, dominant and overdominant pattern. Plant Mol. Biol. 2007, 63, 381–391. [Google Scholar] [CrossRef]
- Stupar, R.M.; Gardiner, J.M.; Oldre, A.G.; Haun, W.J.; Chandler, V.L.; Springer, N.M. Gene expression analyses in maize inbreds and hybrids with varying levels of heterosis. BMC Plant Biol. 2008, 8, 33. [Google Scholar]
- Uzarowska, A.; Keller, B.; Piepho, H.P.; Schwarz, G.; Ingvardsen, C.; Wenzel, G.; Lubberstedt, T. Comparative expression profiling in meristems of inbred-hybrid triplets of maize based on morphological investigations of heterosis for plant height. Plant Mol. Biol. 2007, 63, 21–34. [Google Scholar]
- Sun, Q.; Wu, L.; Ni, Z.; Meng, F.; Wang, Z.; Lin, Z. Differential gene expression patterns in leaves between hybrids and their parental inbreds are correlated with heterosis in a wheat diallel cross. Plant Sci. 2004, 166, 651–657. [Google Scholar] [CrossRef]
- Swanson-Wagner, R.A.; DeCook, R.; Jia, Y.; Bancroft, T.; Ji, T.; Zhao, X.; Nettleton, D.; Schnable, P.S. Paternal dominance of trans-eQTL influences gene expression patterns in maize hybrids. Science 2009, 326, 1118–1120. [Google Scholar]
- Wei, G.; Tao, Y.; Liu, G.; Chen, C.; Luo, R.; Xia, H.; Gan, Q.; Zeng, H.; Lu, Z.; Han, Y.; et al. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc. Natl. Acad. Sci. USA 2009, 106, 7695–7701. [Google Scholar]
- Swanson-Wagner, R.A.; Jia, Y.; DeCook, R.; Borsuk, L.A.; Nettleton, D.; Schnable, P.S. All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc. Natl. Acad. Sci. USA 2006, 103, 6805–6810. [Google Scholar]
- Song, G.S.; Zhai, H.L.; Peng, Y.G.; Zhang, L.; Wei, G.; Chen, X.Y.; Xiao, Y.G.; Wang, L.; Chen, Y.J.; Wu, B.; et al. Comparative transcriptional profiling and preliminary study on heterosis mechanism of super-hybrid rice. Mol. Plant 2010, 3, 1012–1025. [Google Scholar] [CrossRef]
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [CrossRef]
- Ha, M.; Lu, J.; Tian, L.; Ramachandran, V.; Kasschau, K.D.; Chapman, E.J.; Carrington, J.C.; Chen, X.; Wang, X.J.; Chen, Z.J. Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Natl. Acad. Sci. USA 2009, 106, 17835–17840. [Google Scholar]
- He, G.; Zhu, X.; Elling, A.A.; Chen, L.; Wang, X.; Guo, L.; Liang, M.; He, H.; Zhang, H.; Chen, F.; et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 2010, 22, 17–33. [Google Scholar]
- Liu, H.; Qin, C.; Chen, Z.; Zuo, T.; Yang, X.; Zhou, H.; Xu, M.; Cao, S.; Shen, Y.; Lin, H.; et al. Identification of miRNAs and their target genes in developing maize ears by combined small RNA and degradome sequencing. BMC Genomics 2014, 15, 25. [Google Scholar]
- Hollick, J.B.; Chandler, V.L. Epigenetic allelic states of a maize transcriptional regulatory locus exhibit overdominant gene action. Genetics 1998, 150, 891–897. [Google Scholar]
- Zhao, X.; Chai, Y.; Liu, B. Epigenetic inheritance and variation of DNA methylation level and pattern in maize intra-specific hybrids. Plant Sci. 2007, 172, 930–938. [Google Scholar] [CrossRef]
- Ni, Z.; Kim, E.D.; Ha, M.; Lackey, E.; Liu, J.; Zhang, Y.; Sun, Q.; Chen, Z.J. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 2009, 457, 327–331. [Google Scholar]
- Wang, X.; Cao, H.; Zhang, D.; Li, B.; He, Y.; Li, J.; Wang, S. Relationship between differential gene expression and heterosis during ear development in maize (zea mays L.). J. Genet. Genomics 2007, 34, 160–170. [Google Scholar] [CrossRef]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar]
- Li, P.; Ponnala, L.; Gandotra, N.; Wang, L.; Si, Y.; Tausta, S.L.; Kebrom, T.H.; Provart, N.; Patel, R.; Myers, C.R.; et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010, 42, 1060–1067. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 maize genome: Complexity, diversity, and dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology—The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Wu, J.; Mao, X.; Cai, T.; Luo, J.; Wei, L. KOBAS server: A web-based platform for automated annotation and pathway identification. Nucleic Acids Res. 2006, 34, W720–W724. [Google Scholar] [CrossRef]
- Vilella, A.J.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009, 19, 327–335. [Google Scholar]
- Liang, C.; Jaiswal, P.; Hebbard, C.; Avraham, S.; Buckler, E.S.; Casstevens, T.; Hurwitz, B.; McCouch, S.; Ni, J.; Pujar, A.; et al. Gramene: A growing plant comparative genomics resource. Nucleic Acids Res. 2008, 36, D947–D953. [Google Scholar]
- Eveland, A.L.; Satoh-Nagasawa, N.; Goldshmidt, A.; Meyer, S.; Beatty, M.; Sakai, H.; Ware, D.; Jackson, D. Digital gene expression signatures for maize development. Plant Physiol. 2010, 154, 1024–1039. [Google Scholar] [CrossRef]
- Fu, H.; Dooner, H.K. Intraspecific violation of genetic colinearity and its implications in maize. Proc. Natl. Acad. Sci. USA 2002, 99, 9573–9578. [Google Scholar]
- Lai, J.; Li, R.; Xu, X.; Jin, W.; Xu, M.; Zhao, H.; Xiang, Z.; Song, W.; Ying, K.; Zhang, M.; et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat. Genet. 2010, 42, 1027–1030. [Google Scholar]
- Shen, Y.; Zhang, Y.; Chen, J.; Lin, H.; Zhao, M.; Peng, H.; Liu, L.; Yuan, G.; Zhang, S.; Zhang, Z.; et al. Genome expression profile analysis reveals important transcripts in maize roots responding to the stress of heavy metal Pb. Physiol. Plant. 2012, 147, 270–282. [Google Scholar]
- Xiao, S.; Jia, J.; Mo, D.; Wang, Q.; Qin, L.; He, Z.; Zhao, X.; Huang, Y.; Li, A.; Yu, J.; et al. Understanding PRRSV infection in porcine lung based on genome-wide transcriptome response identified by deep sequencing. PLoS One 2010, 5, e11377. [Google Scholar]
- Gibson, G.; Riley-Berger, R.; Harshman, L.; Kopp, A.; Vacha, S.; Nuzhdin, S.; Wayne, M. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics 2004, 167, 1791–1799. [Google Scholar] [CrossRef]
- Charlesworth, D.; Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2009, 10, 783–796. [Google Scholar] [CrossRef]
- Guo, M.; Rupe, M.A.; Zinselmeier, C.; Habben, J.; Bowen, B.A.; Smith, O.S. Allelic variation of gene expression in maize hybrids. Plant Cell 2004, 16, 1707–1716. [Google Scholar]
- Birchler, J.A.; Riddle, N.C.; Auger, D.L.; Veitia, R.A. Dosage balance in gene regulation: Biological implications. Trends Genet. 2005, 21, 219–226. [Google Scholar] [CrossRef]
- Springer, N.M.; Stupar, R.M. Allelic variation and heterosis in maize: How do two halves make more than a whole? Genome Res. 2007, 17, 264–275. [Google Scholar] [CrossRef]
- Zhu, Y.; Fu, J.; Zhang, J.; Liu, T.; Jia, Z.; Wang, J.; Jin, Y.; Lian, Y.; Wang, M.; Zheng, J.; et al. Genome-wide analysis of gene expression profiles during ear development of maize. Plant Mol. Biol. 2009, 70, 63–77. [Google Scholar] [CrossRef]
- Qin, Y.F.; Fang, H.M.; Tian, Q.N.; Bao, Z.X.; Lu, P.; Zhao, J.M.; Mai, J.; Zhu, Z.Y.; Shu, L.L.; Zhao, L.; et al. Transcriptome profiling and digital gene expression by deep-sequencing in normal/regenerative tissues of planarian Dugesia japonica. Genomics 2011, 97, 364–371. [Google Scholar] [CrossRef]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆Ct method. Methods 2001, 25, 402–408. [Google Scholar]
- C’t Hoen, P.A.; Ariyurek, Y.; Thygesen, H.H.; Vreugdenhil, E.; Vossen, R.H.; de Menezes, R.X.; Boer, J.M.; van Ommen, G.J.B.; den Dunnen, J.T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Res. 2008, 36, e141. [Google Scholar] [CrossRef]
- Morrissy, A.S.; Morin, R.D.; Delaney, A.; Zeng, T.; McDonald, H.; Jones, S.; Zhao, Y.; Hirst, M.; Marra, M.A. Next-generation tag sequencing for cancer gene expression profiling. Genome Res. 2009, 19, 1825–1835. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar]
- Consortium, T.G.O. The gene ontology project in 2008. Nucleic Acids Res. 2008, 36, D440–D444. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG Database. In “In Silico”Simulation of Biological Processes; John Wiley & Sons: Manhattan, NY, USA, 2008; pp. 91–103. [Google Scholar]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ding, H.; Qin, C.; Luo, X.; Li, L.; Chen, Z.; Liu, H.; Gao, J.; Lin, H.; Shen, Y.; Zhao, M.; et al. Heterosis in Early Maize Ear Inflorescence Development: A Genome-Wide Transcription Analysis for Two Maize Inbred Lines and Their Hybrid. Int. J. Mol. Sci. 2014, 15, 13892-13915. https://doi.org/10.3390/ijms150813892
Ding H, Qin C, Luo X, Li L, Chen Z, Liu H, Gao J, Lin H, Shen Y, Zhao M, et al. Heterosis in Early Maize Ear Inflorescence Development: A Genome-Wide Transcription Analysis for Two Maize Inbred Lines and Their Hybrid. International Journal of Molecular Sciences. 2014; 15(8):13892-13915. https://doi.org/10.3390/ijms150813892
Chicago/Turabian StyleDing, Haiping, Cheng Qin, Xirong Luo, Lujiang Li, Zhe Chen, Hongjun Liu, Jian Gao, Haijian Lin, Yaou Shen, Maojun Zhao, and et al. 2014. "Heterosis in Early Maize Ear Inflorescence Development: A Genome-Wide Transcription Analysis for Two Maize Inbred Lines and Their Hybrid" International Journal of Molecular Sciences 15, no. 8: 13892-13915. https://doi.org/10.3390/ijms150813892