Effect of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) on Hormones of Energy Balance in a TCDD-Sensitive and a TCDD-Resistant Rat Strain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Body Weight Change

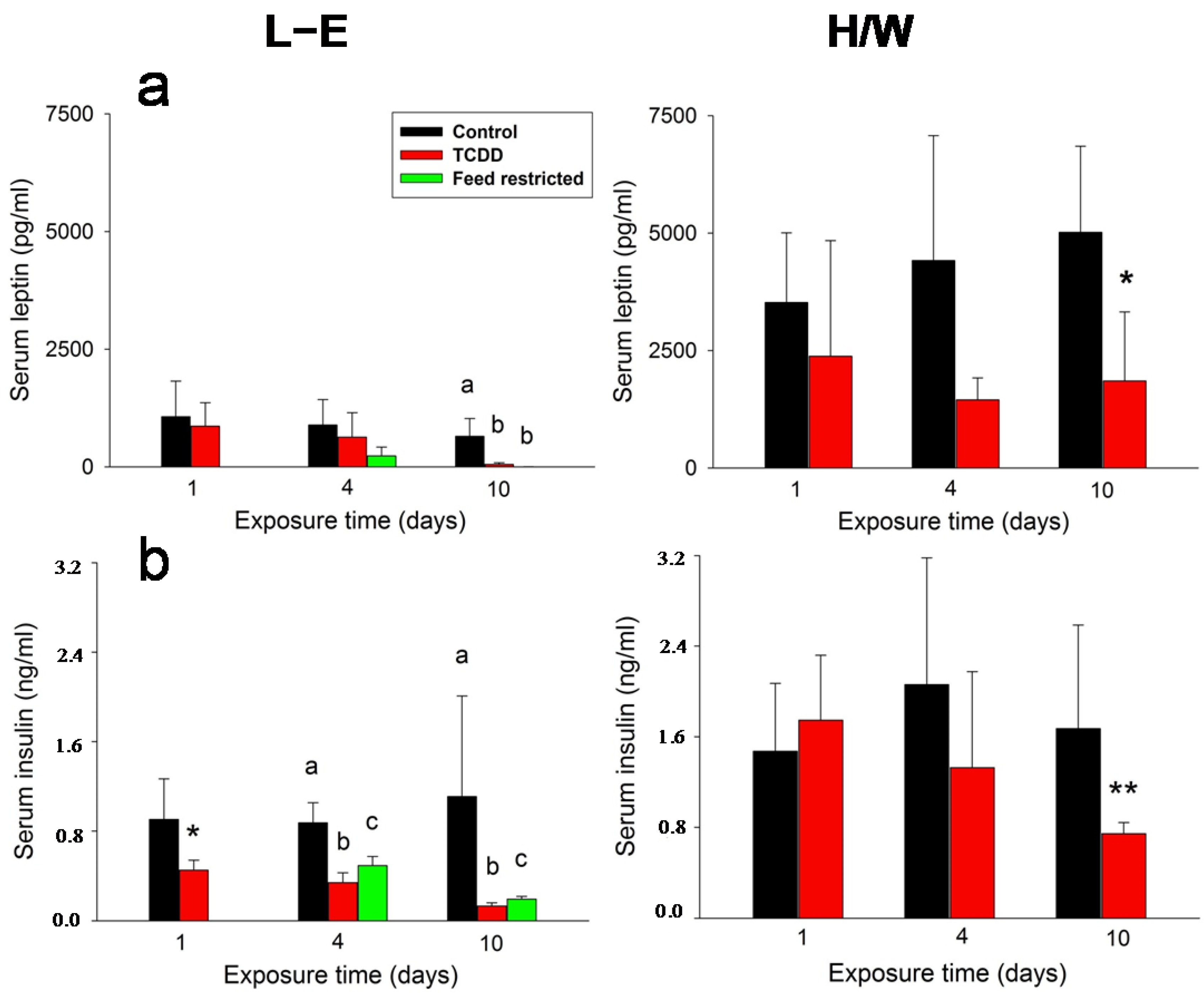
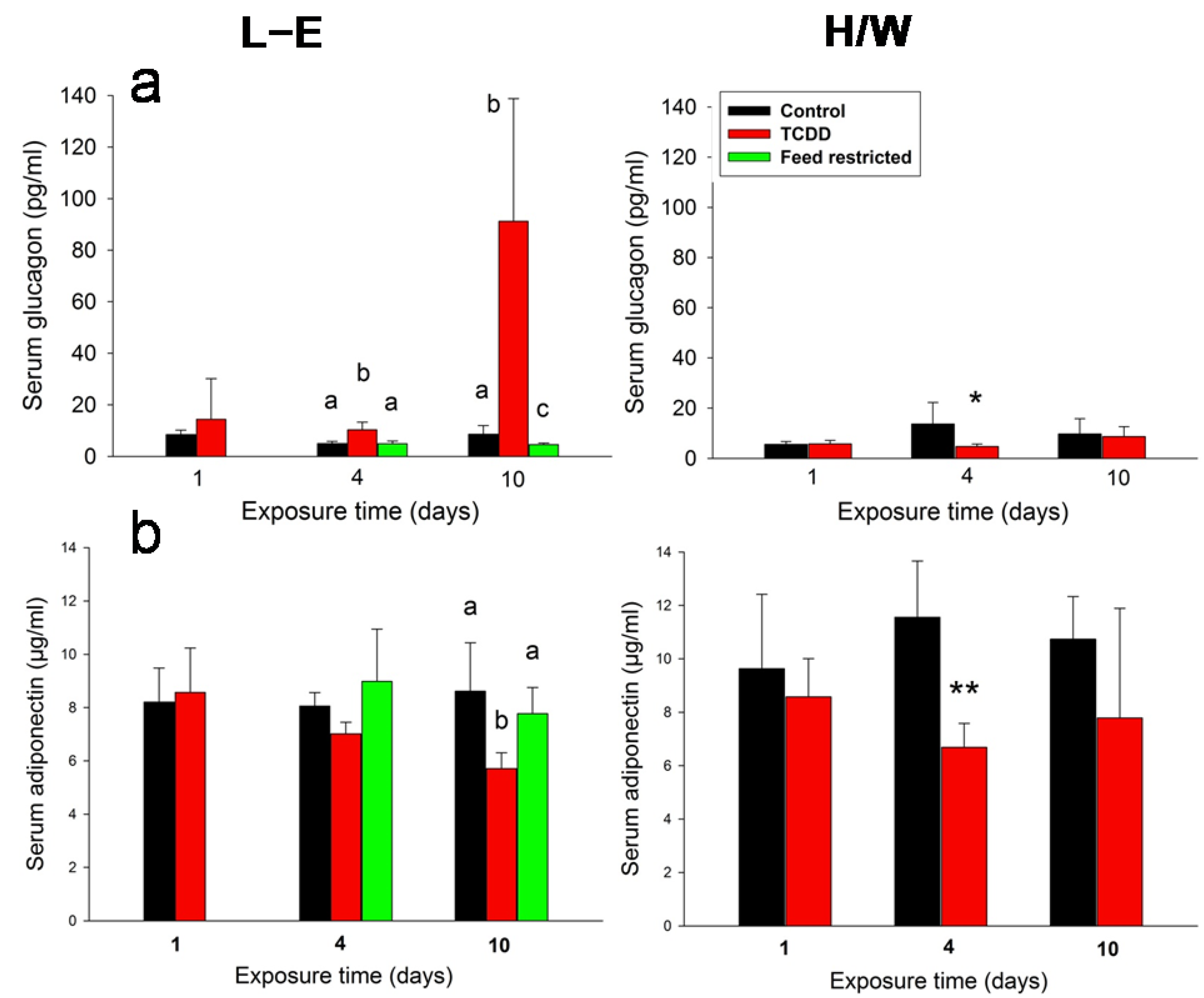
2.2. Leptin, Insulin, Glucagon, and Adiponectin Serum Concentrations

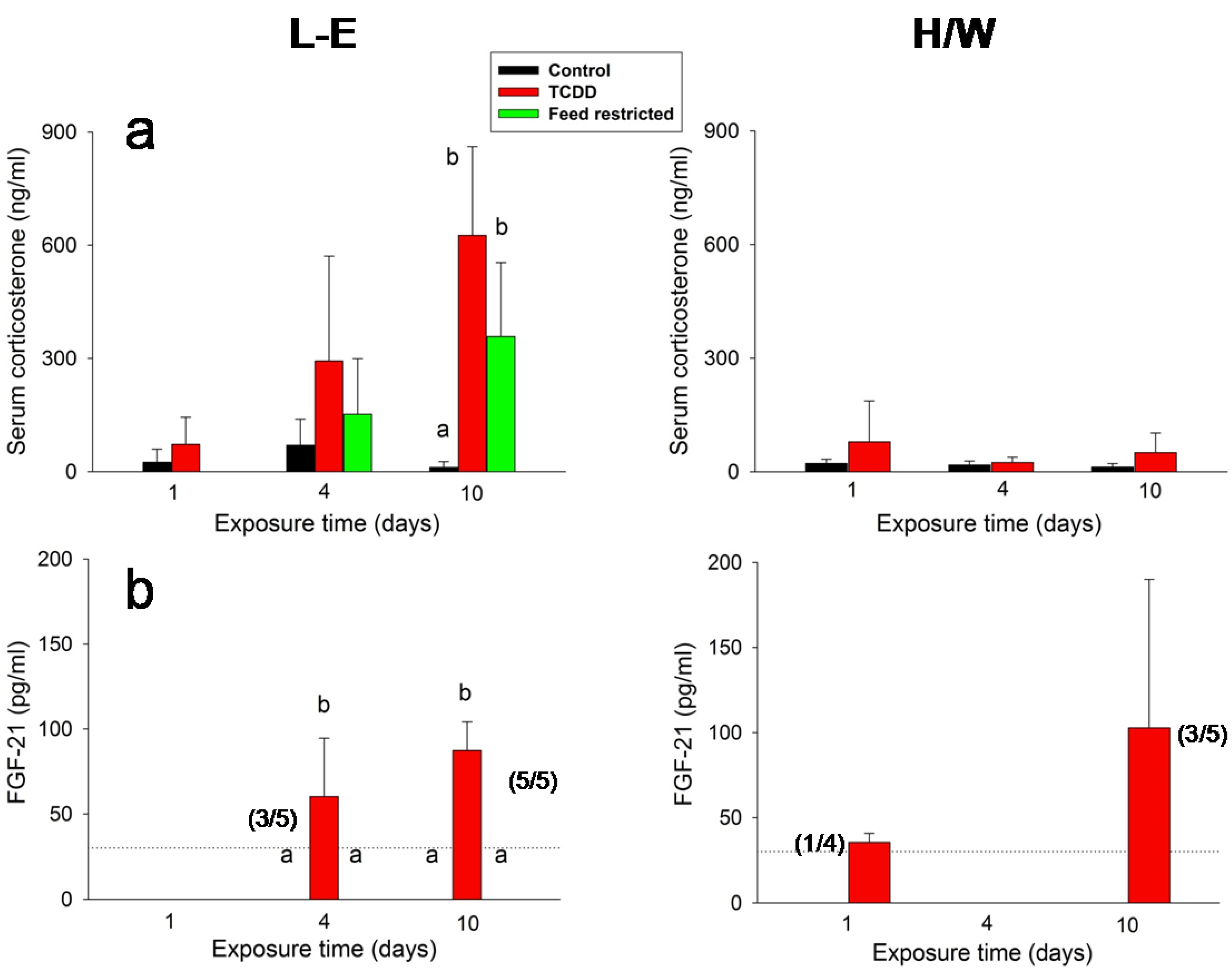
2.3. IGF-1, Ghrelin, Corticosterone and FGF-21 Serum Concentrations

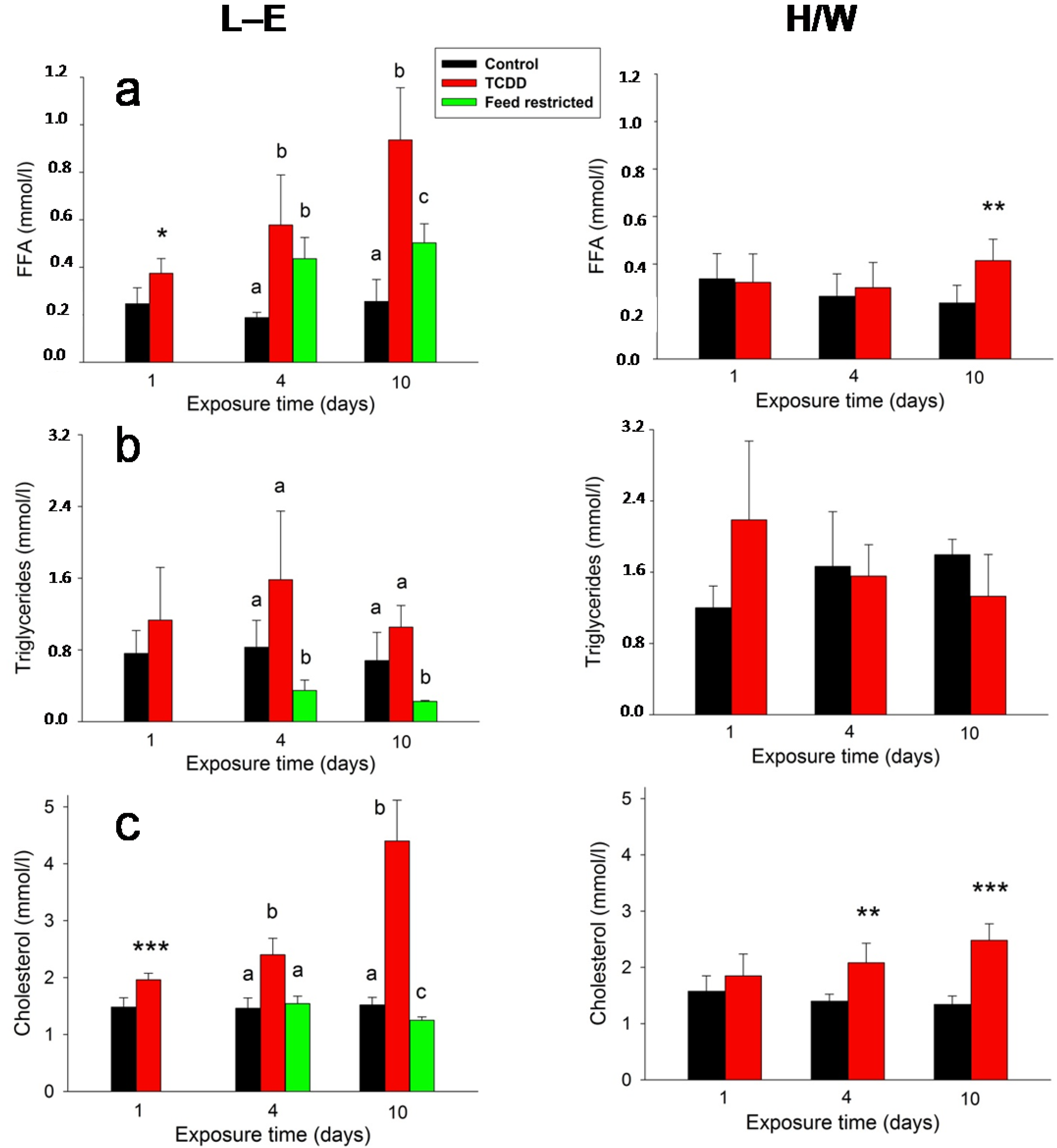
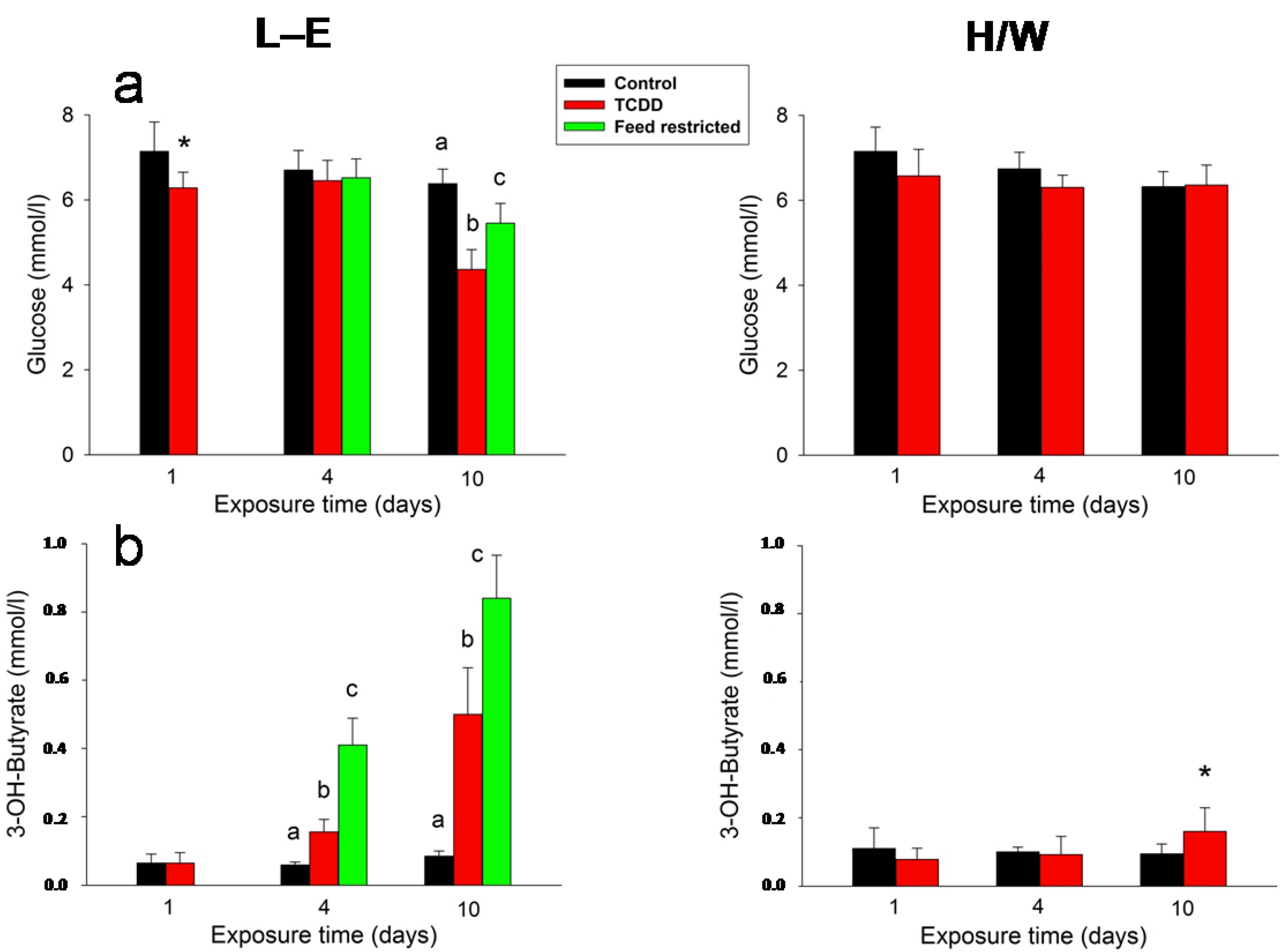
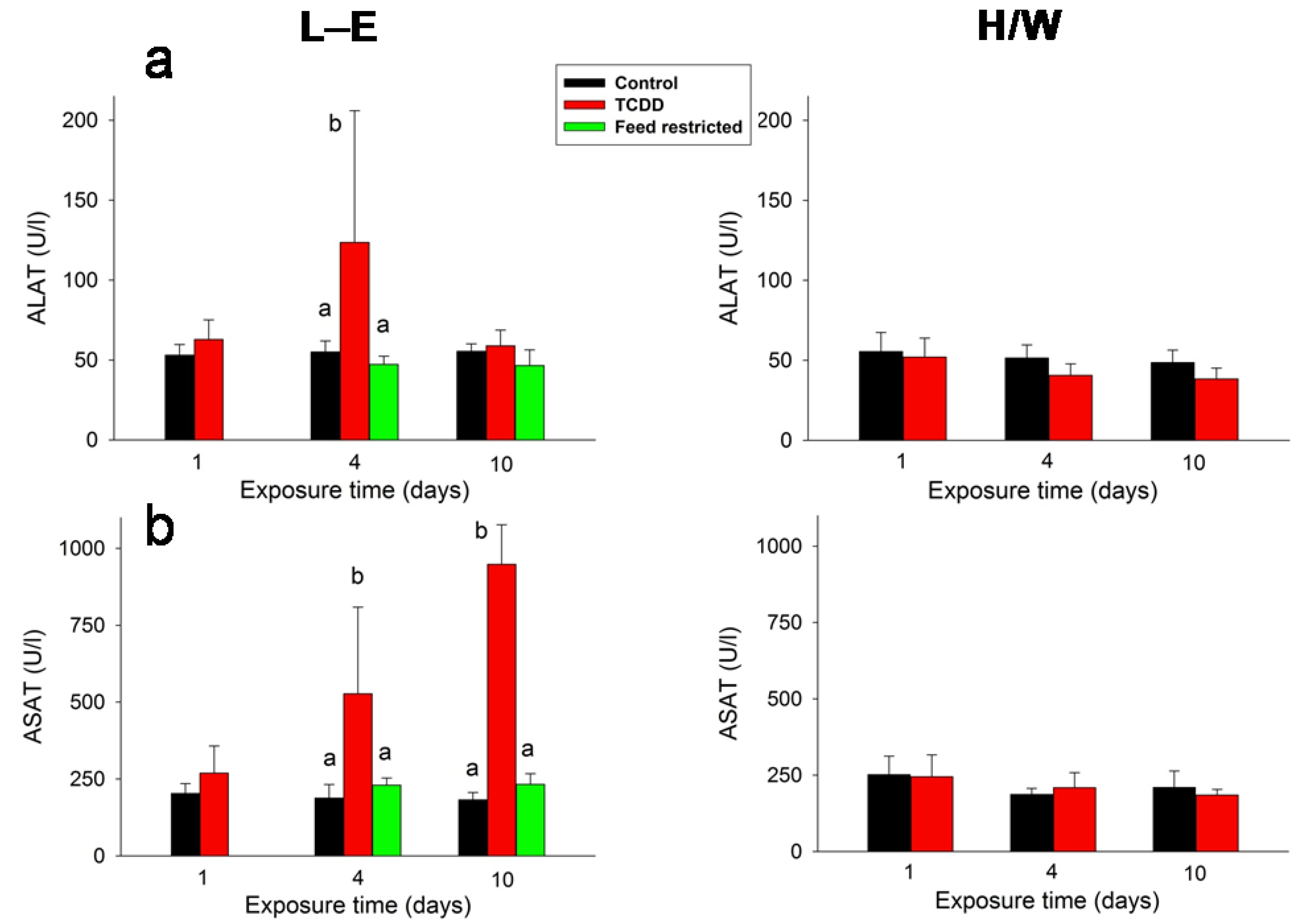
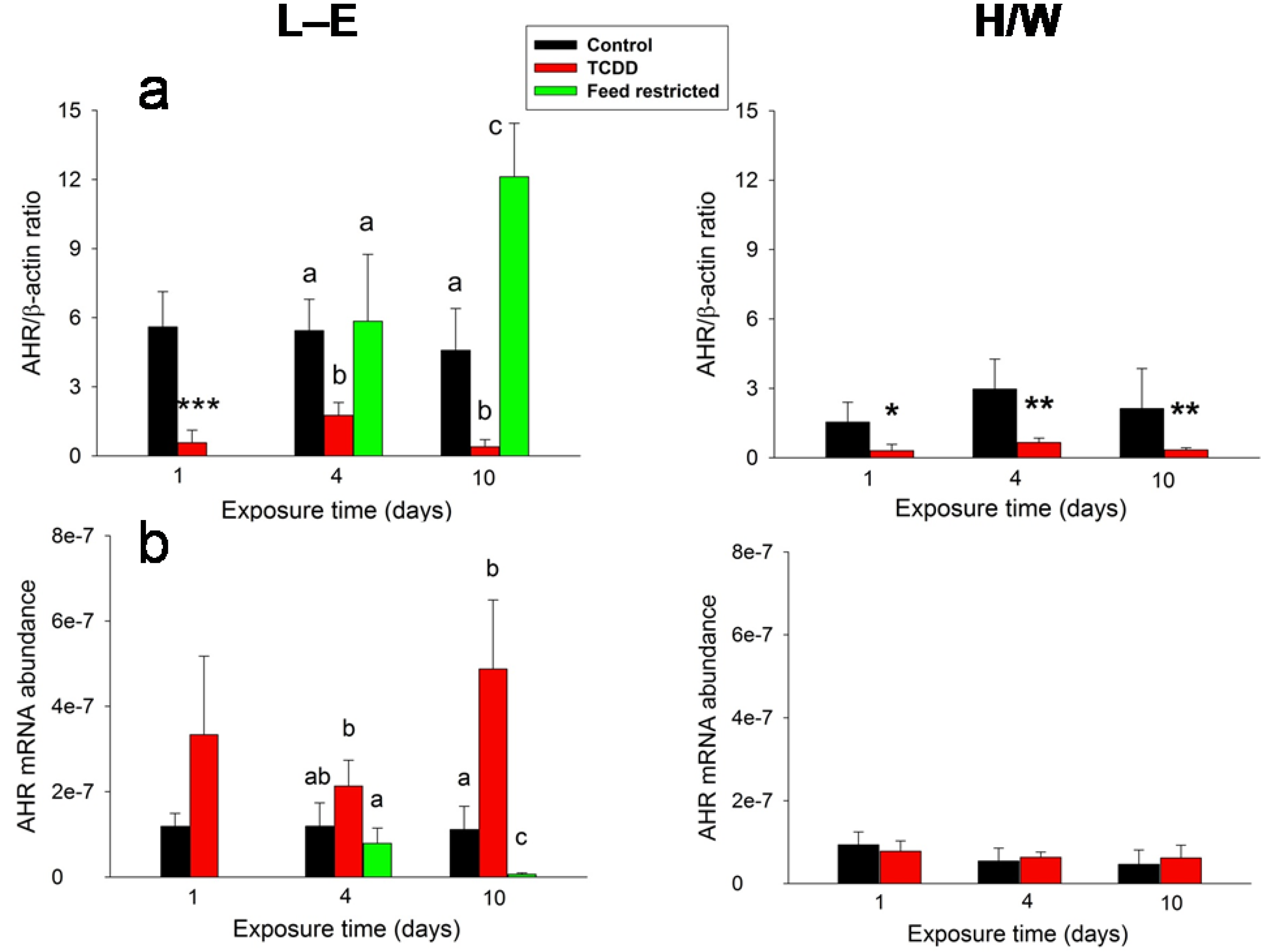
2.4. Clinical Chemistry and AHR



2.5. General Discussion


2.5.1. Insulin and Leptin

2.5.2. Ghrelin and Corticosterone
2.5.3. Glugacon, Glucose and Adiponectin
2.5.4. IGF-1and FGF-21
2.5.5. Clinical Chemistry and AHR
3. Experimental Section
3.1. Animals and Husbandry
3.2. Experimental Design
3.3. Peptide Assays
3.4. Clinical Chemistry and AHR Analysis
3.5. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pohjanvirta, R.; Tuomisto, J. Short-term toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in laboratory animals: Effects, mechanisms, and animal models. Pharmacol. Rev. 1994, 46, 483–549. [Google Scholar]
- Birnbaum, L.S.; Tuomisto, J. Non-carcinogenic effects of TCDD in animals. Food Addit. Contam. 2000, 17, 275–288. [Google Scholar] [CrossRef]
- Abbott, B.D. Teratogenic impact of dioxin activated AHR in laboratory animals. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2012; pp. 257–266. [Google Scholar]
- Mocarelli, P.; Gerthoux, P.M.; Needham, L.L.; Patterson, D.G.; Limonta, G.; Falbo, R.; Signorini, S.; Bertona, M.; Crespi, C.; Sarto, C.; et al. Perinatal exposure to low doses of dioxin can permanently impair human semen quality. Environ. Health Perspect. 2011, 119, 713–718. [Google Scholar] [CrossRef]
- Alaluusua, S.; Lukinmaa, P.L. Developmental dental toxicity of dioxin and related compounds—A review. Int. Dent. J. 2006, 56, 323–331. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Korkalainen, M.; Moffat, I.D.; Boutros, P.C.; Okey, A.B. Role of the AHR and its structure in TCDD toxicity. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Pohjanvirta, R. Transgenic mouse lines expressing rat AH receptor variants—A new animal model for research on AH receptor function and dioxin toxicity mechanisms. Toxicol. Appl. Pharmacol. 2009, 236, 166–182. [Google Scholar] [CrossRef]
- Lindén, J.; Lensu, S.; Tuomisto, J.; Pohjanvirta, R. Dioxins, the aryl hydrocarbon receptor and the central regulation of energy balance. Front. Neuroendocrinol. 2010, 31, 452–478. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Miettinen, H.M.; Sankari, S.; Hegde, N.; Lindén, J. Unexpected gender difference in sensitivity to the acute toxicity of dioxin in mice. Toxicol. Appl. Pharmacol. 2012, 262, 167–176. [Google Scholar] [CrossRef]
- Gasiewicz, T.A.; Neal, R.A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin tissue distribution, excretion, and effects on clinical chemical parameters in guinea pigs. Toxicol. Appl. Pharmacol. 1979, 51, 329–339. [Google Scholar] [CrossRef]
- Henry, E.C.; Gasiewicz, T.A. Changes in thyroid hormones and thyroxine glucuronidation in hamsters compared with rats following treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 1987, 89, 165–174. [Google Scholar] [CrossRef]
- Kelling, C.K.; Christian, B.J.; Inhorn, S.L.; Peterson, R.E. Hypophagia-induced weight loss in mice, rats, and guinea pigs treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam. Appl. Toxicol. 1985, 5, 700–712. [Google Scholar] [CrossRef]
- Olson, J.R.; Holscher, M.A.; Neal, R.A. Toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the golden Syrian hamster. Toxicol. Appl. Pharmacol. 1980, 55, 67–78. [Google Scholar] [CrossRef]
- Geusau, A.; Schmaldienst, S.; Derfler, K.; Päpke, O.; Abraham, K. Severe 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) intoxication: Kinetics and trials to enhance elimination in two patients. Arch. Toxicol. 2002, 76, 316–325. [Google Scholar] [CrossRef]
- Needham, L.L.; Gerthoux, P.M.; Patterson, D.G.; Brambilla, P.; Turner, W.E.; Beretta, C.; Pirkle, J.L.; Colombo, L.; Sampson, E.J.; Tramacere, P.; et al. Serum dioxin levels in Seveso, Italy, population in 1976. Teratog. Carcinog. Mutagen. 1997, 17, 225–240. [Google Scholar]
- Sorg, O.; Zennegg, M.; Schmid, P.; Fedosyuk, R.; Valikhnovskyi, R.; Gaide, O.; Kniazevych, V.; Saurat, J.H. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: Identification and measurement of TCDD metabolites. Lancet 2009, 374, 1179–1185. [Google Scholar] [CrossRef]
- Schneeberger, M.; Gomis, R.; Claret, M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 2014, 220, T25–T46. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Woods, S.C.; Lutz, T.A.; Geary, N.; Langhans, W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos. Trans. R. Soc. B: Biol. Sci. 2006, 361, 1219–1235. [Google Scholar] [CrossRef]
- Guo, S. Insulin signaling, resistance, and metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, T1–T23. [Google Scholar] [CrossRef]
- Gerozissis, K. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur. J. Pharmacol. 2008, 585, 38–49. [Google Scholar] [CrossRef]
- Sánchez-Lasheras, C.; Könner, A.C.; Brüning, J.C. Integrative neurobiology of energy homeostasis-neurocircuits, signals and mediators. Front. Neuroendocrinol. 2010, 31, 4–15. [Google Scholar] [CrossRef]
- Grill, H.J.; Schwartz, M.W.; Kaplan, J.M.; Foxhall, J.S.; Breininger, J.; Baskin, D.G. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology 2002, 143, 239–246. [Google Scholar]
- Halaas, J.L.; Boozer, C.N.; Blair-West, J.; Fidahusein, N.; Denton, D.A.; Friedman, J.M. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc. Natl. Acad. Sci. USA 1997, 94, 8878–8883. [Google Scholar]
- Könner, A.C.; Klöckener, T.; Brüning, J.C. Control of energy homeostasis by insulin and leptin: Targeting the arcuate nucleus and beyond. Physiol. Behav. 2009, 97, 632–638. [Google Scholar] [CrossRef]
- Sahu, A. Effects of chronic central leptin infusion on proopiomelanocortin and neurotensin gene expression in the rat hypothalamus. Neurosci. Lett. 2008, 440, 125–129. [Google Scholar] [CrossRef]
- Filippi, B.M.; Abraham, M.A.; Yue, J.T.Y.; Lam, T.K.T. Insulin and glucagon signaling in the central nervous system. Rev. Endocr. Metab. Disord. 2013, 14, 365–375. [Google Scholar] [CrossRef]
- Berthoud, H.R. Vagal and hormonal gut-brain communication: From satiation to satisfaction. Neurogastroenterol. Motil. 2008, 20, 64–72. [Google Scholar] [CrossRef]
- Dietrich, M.O.; Horvath, T.L. Feeding signals and brain circuitry. Eur. J. Neurosci. 2009, 30, 1688–1696. [Google Scholar] [CrossRef]
- Pusztai, P.; Sarman, B.; Ruzicska, E.; Toke, J.; Racz, K.; Somogyi, A.; Tulassay, Z. Ghrelin: A new peptide regulating the neurohormonal system, energy homeostasis and glucose metabolism. Diabetes Metab. Res. Rev. 2008, 24, 343–352. [Google Scholar] [CrossRef]
- Schellekens, H.; Finger, B.C.; Dinan, T.G.; Cryan, J.F. Ghrelin signalling and obesity: At the interface of stress, mood and food reward. Pharmacol. Ther. 2012, 135, 316–326. [Google Scholar] [CrossRef]
- Date, Y.; Kojima, M.; Hosoda, H.; Sawaguchi, A.; Mondal, M.S.; Suganuma, T.; Matsukura, S.; Kangawa, K.; Nakazato, M. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 2000, 141, 4255–4261. [Google Scholar]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Kageyama, H.; Takenoya, F.; Shiba, K.; Shioda, S. Neuronal circuits involving ghrelin in the hypothalamus-mediated regulation of feeding. Neuropeptides 2010, 44, 133–138. [Google Scholar] [CrossRef]
- Begg, D.P.; Woods, S.C. The endocrinology of food intake. Nat. Rev. Endocrinol. 2013, 9, 584–597. [Google Scholar] [CrossRef]
- Alsahli, M.; Gerich, J.E. Hypoglycemia. Endocrinol. Metab. Clin. N. Am. 2013, 42, 657–676. [Google Scholar] [CrossRef]
- Ramnanan, C.J.; Edgerton, D.S.; Kraft, G.; Cherrington, A.D. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes. Metab. 2011, 13, 118–125. [Google Scholar]
- Cryer, P.E. Minireview: Glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology 2012, 153, 1039–1048. [Google Scholar] [CrossRef]
- Parker, J.A.; McCullough, K.A.; Field, B.C.T.; Minnion, J.S.; Martin, N.M.; Ghatei, M.A.; Bloom, S.R. Glucagon and GLP-1 inhibit food intake and increase c-fos expression in similar appetite regulating centres in the brainstem and amygdala. Int. J. Obes. 2013, 37, 1391–1398. [Google Scholar] [CrossRef]
- Tishinsky, J.M.; Robinson, L.E.; Dyck, D.J. Insulin-sensitizing properties of adiponectin. Biochimie 2012, 94, 2131–2136. [Google Scholar] [CrossRef]
- Kubota, N.; Yano, W.; Kubota, T.; Yamauchi, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Okamoto, S.; Shiuchi, T.; et al. Adiponectin Stimulates AMP-Activated Protein Kinase in the Hypothalamus and Increases Food Intake. Cell Metab. 2007, 6, 55–68. [Google Scholar]
- Bassi, M.; Do Carmo, J.M.; Hall, J.E.; Da Silva, A.A. Chronic effects of centrally administered adiponectin on appetite, metabolism and blood pressure regulation in normotensive and hypertensive rats. Peptides 2012, 37, 1–5. [Google Scholar] [CrossRef]
- Rose, A.J.; Herzig, S. Metabolic control through glucocorticoid hormones: An update. Mol. Cell. Endocrinol. 2013, 380, 65–78. [Google Scholar] [CrossRef]
- Rose, A.J.; Vegiopoulos, A.; Herzig, S. Role of glucocorticoids and the glucocorticoid receptor in metabolism: Insights from genetic manipulations. J. Steroid Biochem. Mol. Biol. 2010, 122, 10–20. [Google Scholar] [CrossRef]
- Stanley, B.G.; Lanthier, D.; Chin, A.S.; Leibowitz, S.F. Suppression of neuropeptide Y-elicited eating by adrenalectomy or hypophysectomy: Reversal with corticosterone. Brain Res. 1989, 501, 32–36. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta—Gene Struct. Expr. 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Izumiya, Y.; Bina, H.A.; Ouchi, N.; Akasaki, Y.; Kharitonenkov, A.; Walsh, K. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008, 582, 3805–3810. [Google Scholar] [CrossRef]
- Tacer, K.F.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 2010, 24, 2050–2064. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef]
- Woo, Y.C.; Xu, A.; Wang, Y.; Lam, K.S.L. Fibroblast Growth Factor 21 as an emerging metabolic regulator: Clinical perspectives. Clin. Endocrinol. 2013, 78, 489–496. [Google Scholar] [CrossRef]
- Badman, M.K.; Koester, A.; Flier, J.S.; Kharitonenkov, A.; Maratos-Flier, E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology 2009, 150, 4931–4940. [Google Scholar] [CrossRef]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine Regulation of the Fasting Response by PPAR-α-Mediated Induction of Fibroblast Growth Factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.-S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef]
- Zhang, X.; Yeung, D.C.Y.; Karpisek, M.; Stejskal, D.; Zhou, Z.-G.; Liu, F.; Wong, R.L.C.; Chow, W.-S.; Tso, A.W.K.; Lam, K.S.L.; et al. Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans. Diabetes 2008, 57, 1246–1253. [Google Scholar] [CrossRef]
- Sjögren, K.; Liu, J.-L.; Blad, K.; Skrtic, S.; Vidal, O.; Wallenius, V.; Leroith, D.; Törnell, J.; Isaksson, O.G.P.; Jansson, J.-O.; et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7088–7092. [Google Scholar] [CrossRef]
- List, E.O.; Sackmann-Sala, L.; Berryman, D.E.; Funk, K.; Kelder, B.; Gosney, E.S.; Okada, S.; Ding, J.; Cruz-Topete, D.; Kopchick, J.J. Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR-/-) mouse. Endocr. Rev. 2011, 32, 356–386. [Google Scholar] [CrossRef]
- Yakar, S.; Adamo, M.L. Insulin-like growth factor 1 physiology. Lessons from mouse models. Endocrinol. Metab. Clin. N. Am. 2012, 41, 231–247. [Google Scholar] [CrossRef]
- Inagaki, T.; Lin, V.Y.; Goetz, R.; Mohammadi, M.; Mangelsdorf, D.J.; Kliewer, S.A. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 2008, 8, 77–83. [Google Scholar] [CrossRef]
- Fazeli, P.K.; Klibanski, A. Determinants of GH resistance in malnutrition. J. Endocrinol. 2014, 220, R57–R65. [Google Scholar] [CrossRef]
- Lam, C.K.L.; Chari, M.; Lam, T.K.T. CNS regulation of glucose homeostasis. Physiology 2009, 24, 159–170. [Google Scholar] [CrossRef]
- Thorens, B. Brain glucose sensing and neural regulation of insulin and glucagon secretion. Diabetes Obes. Metab. 2011, 13, 82–88. [Google Scholar] [CrossRef]
- Dunn-Meynell, A.A.; Sanders, N.M.; Compton, D.; Becker, T.C.; Eiki, J.I.; Zhang, B.B.; Levin, B.E. Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. J. Neurosci. 2009, 29, 7015–7022. [Google Scholar]
- Lam, T.K.T. Neuronal regulation of homeostasis by nutrient sensing. Nat. Med. 2010, 16, 392–395. [Google Scholar] [CrossRef]
- Burdakov, D.; Karnani, M.M.; Gonzalez, A. Lateral hypothalamus as a sensor-regulator in respiratory and metabolic control. Physiol. Behav. 2013, 121, 117–124. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef]
- Bestervelt, L.L.; Pitt, J.A.; Nolan, C.J.; Piper, W.N. TCDD alters pituitary-adrenal function II: Evidence for decreased bioactivity of ACTH. Neurotoxicol. Teratol. 1993, 15, 371–376. [Google Scholar] [CrossRef]
- Bestervelt, L.L.; Pitt, J.A.; Piper, W.N. Evidence for ah receptor mediation of increased ACTH concentrations in primary cultures of rat anterior pituitary cells exposed to TCDD. Toxicol. Sci. 1998, 46, 294–299. [Google Scholar] [CrossRef]
- Schuur, A.G.; Boekhorst, F.M.; Brouwer, A.; Visser, T.J. Extrathyroidal effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on thyroid hormone turnover in male Sprague-Dawley rats. Endocrinology 1997, 138, 3727–3734. [Google Scholar]
- Pohjanvirta, R.; Laitinen, J.; Vakkuri, O.; Lindén, J.; Kokkola, T.; Unkila, M.; Tuomisto, J. Mechanism by which 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) reduces circulating melatonin levels in the rat. Toxicology 1996, 107, 85–97. [Google Scholar] [CrossRef]
- DiBartolemeis, M.J.; Moore, R.W.; Peterson, R.E.; Jefcoate, C.R. Hypercholesterolemia and the regulation of adrenal steroidogenesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. Toxicol. Appl. Pharmacol. 1986, 85, 313–323. [Google Scholar] [CrossRef]
- Moore, R.W.; Jefcoate, C.R.; Peterson, R.E. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibits steroidogenesis in the rat testis by inhibiting the mobilization of cholesterol to cytochrome P450scc. Toxicol. Appl. Pharmacol. 1991, 109, 85–97. [Google Scholar] [CrossRef]
- Brunnberg, S.; Svendebrog, E.; Gustafsson, J.Å. Functional interactions of AHR with other receptors. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Gorski, J.R.; Muzi, G.; Weber, L.W.; Pereira, D.W.; Arceo, R.J.; Iatropoulos, M.J.; Rozman, K. Some endocrine and morphological aspects of the acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Pathol. 1988, 16, 313–320. [Google Scholar] [CrossRef]
- Gorski, J.R.; Rozman, K. Dose-response and time course of hypothyroxinemia and hypoinsulinemia and characterization of insulin hypersensitivity in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Toxicology 1987, 44, 297–307. [Google Scholar] [CrossRef]
- Novelli, M.; Piaggi, S.; de Tata, V. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced impairment of glucose-stimulated insulin secretion in isolated rat pancreatic islets. Toxicol. Lett. 2005, 156, 307–314. [Google Scholar] [CrossRef]
- Tuomisto, J.T.; Laaksonen, M.; Viluksela, M.; Pohjanvirta, R.; Tuomisto, J. Minor changes in leptin levels after 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) exposure. Pharmacol. Toxicol. 1997, 80, 113–114. [Google Scholar]
- Bertile, F.; Raclot, T. The melanocortin system during fasting. Peptides 2006, 27, 291–300. [Google Scholar] [CrossRef]
- Johansson, A.; Fredriksson, R.; Winnergren, S.; Hulting, A.-L.; Schiöth, H.B.; Lindblom, J. The relative impact of chronic food restriction and acute food deprivation on plasma hormone levels and hypothalamic neuropeptide expression. Peptides 2008, 29, 1588–1595. [Google Scholar] [CrossRef]
- Lensu, S.; Tiittanen, P.; Pohjanvirta, R. Circadian differences between two rat strains in their feeding and drinking micro- and macrostructures. Biol. Rhythm Res. 2011, 42, 385–405. [Google Scholar] [CrossRef]
- Cone, J.J.; McCutcheon, J.E.; Roitman, M.F. Ghrelin acts as an interface between physiological state and phasic dopamine signaling. J. Neurosci. 2014, 34, 4905–4913. [Google Scholar] [CrossRef]
- Lindén, J.; Korkalainen, M.; Lensu, S.; Tuomisto, J.; Pohjanvirta, R. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and leptin on hypothalamic mRNA expression of factors participating in food intake regulation in a TCDD-sensitive and a TCDD-resistant rat strain. J. Biochem. Mol. Toxicol. 2005, 19, 139–148. [Google Scholar] [CrossRef]
- Tuomisto, J.; Andrzejewski, W.; Unkila, M.; Pohjanvirta, R.; Lindén, J.; Vartiainen, T.; Tuomisto, L. Modulation of TCDD-induced wasting syndrome by portocaval anastomosis and vagotomy in Long-Evans and Han/Wistar rats. Eur. J. Pharmacol. 1995, 292, 277–285. [Google Scholar]
- Moon, B.H.; Hong, C.G.; Kim, S.Y.; Kim, H.J.; Shin, S.K.; Kang, S.; Lee, K.J.; Kim, Y.K.; Lee, M.S.; Shin, K.H. A single administration of 2,3,7,8-tetrachlorodibenzo-p-dioxin that produces reduced food and water intake induces long-lasting expression of corticotropin-releasing factor, arginine vasopressin, and proopiomelanocortin in rat brain. Toxicol. Appl. Pharmacol. 2008, 233, 314–322. [Google Scholar] [CrossRef]
- Viluksela, M.; Unkila, M.; Pohjanvirta, R.; Tuomisto, J.T.; Stahl, B.U.; Rozman, K.K.; Tuomisto, J. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on liver phosphoenolpyruvate carboxykinase (PEPCK) activity, glucose homeostasis and plasma amino acid concentrations in the most TCDD-susceptible and the most TCDD-resistant rat strains. Arch. Toxicol. 1999, 73, 323–336. [Google Scholar] [CrossRef]
- Stahl, B.U.; Beer, D.G.; Weber, L.W.D.; Rozman, K. Reduction of hepatic phosphoenolpyruvate carboxykinase (PEPCK) activity by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) is due to decreased mRNA levels. Toxicology 1993, 79, 81–95. [Google Scholar] [CrossRef]
- Oh, K.-J.; Han, H.-S.; Kim, M.-J.; Koo, S.-H. Transcriptional regulators of hepatic gluconeogenesis. Arch. Pharm. Res. 2013, 36, 189–200. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Tuomisto, J. Remarkable residual alterations in responses to feeding regulatory challenges in Han/Wistar rats after recovery from the acute toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Food Chem. Toxicol. 1990, 28, 677–686. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Unkila, M.; Tuomisto, J. The loss of glucoprivic feeding is an early-stage alteration in TCDD-treated Han/Wistar rats. Pharmacol. Toxicol. 1990, 67, 441–443. [Google Scholar] [CrossRef]
- Hectors, T.L.M.; Vanparys, C.; van Gaal, L.F.; Jorens, P.G.; Covaci, A.; Blust, R. Insulin resistance and environmental pollutants: Experimental evidence and future perspectives. Environ. Health Perspect. 2013, 121, 1273–1281. [Google Scholar]
- Kern, P.A.; Said, S.; Jackson, W.G., Jr.; Michalek, J.E. Insulin sensitivity following Agent Orange exposure in Vietnam veterans with high blood levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Clin. Endocrinol. Metab. 2004, 89, 4665–4672. [Google Scholar] [CrossRef]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Croutch, C.R.; Lebofsky, M.; Schramm, K.-W.; Terranova, P.F.; Rozman, K.K. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) and 1,2,3,4,7,8-hexachlorodibenzo-p-dioxin (HxCDD) alter body weight by decreasing insulin-like growth factor I (IGF-I) signaling. Toxicol. Sci. 2005, 85, 560–571. [Google Scholar] [CrossRef]
- List, E.O.; Berryman, D.E.; Funk, K.; Jara, A.; Kelder, B.; Wang, F.; Stout, M.B.; Zhi, X.; Sun, L.; White, T.A.; et al. Liver-specific GH receptor gene disrupted (LiGHRKO) mice have decreased endocrine IGF-1, increased local IGF-1 as well as altered body size, body composition and adipokine profiles. Endocrinology 2014, 155, 1793–1805. [Google Scholar] [CrossRef]
- Moore, R.W.; Parsons, J.A.; Bookstaff, R.C.; Peterson, R.E. Plasma concentrations of pituitary hormones in 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated male rats. J. Biochem. Toxicol. 1989, 4, 165–172. [Google Scholar] [CrossRef]
- Elango, A.; Shepherd, B.; Chen, T.T. Effects of endocrine disrupters on the expression of growth hormone and prolactin mRNA in the rainbow trout pituitary. Gen. Comp. Endocrinol. 2006, 145, 116–127. [Google Scholar] [CrossRef]
- Boutros, P.C.; Yao, C.Q.; Watson, J.D.; Wu, A.H.; Moffat, I.D.; Prokopec, S.D.; Smith, A.B.; Okey, A.B.; Pohjanvirta, R. Hepatic transcriptomic responses to TCDD in dioxin-sensitive and dioxin-resistant rats during the onset of toxicity. Toxicol. Appl. Pharmacol. 2011, 251, 119–129. [Google Scholar] [CrossRef]
- Clemmons, D.R.; Underwood, L.E. Nutritional regulation of IGF-I and IGF binding proteins. Annu. Rev. Nutr. 1991, 11, 393–412. [Google Scholar] [CrossRef]
- Sharma, N.; Castorena, C.M.; Cartee, G.D. Greater insulin sensitivity in calorie restricted rats occurs with unaltered circulating levels of several important myokines and cytokines. Nutr. Metab. 2012, 9, 90. [Google Scholar] [CrossRef]
- Lu, H.; Cui, W.; Klaassen, C.D. Nrf2 protects against 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-indsuced oxidative injury and steatohepatitis. Toxicol. Appl. Pharmacol. 2011, 256, 122–135. [Google Scholar] [CrossRef]
- Shaban, Z.; El-Shazly, S.; Abdelhady, S.; Fattouh, I.; Muzandu, K.; Ishizuka, M.; Kimura, K.; Kazusaka, A.; Fujita, S. Down regulation of hepatic PPARalpha function by AhR ligand. J. Vet. Med. Sci. 2004, 66, 1377–1386. [Google Scholar] [CrossRef]
- Berglund, E.D.; Kang, L.; Lee-Young, R.S.; Hasenour, C.M.; Lustig, D.G.; Lynes, S.E.; Donahue, E.P.; Swift, L.L.; Charron, M.J.; Wasserman, D.H. Glucagon and lipid interactions in the regulation of hepatic AMPK signaling and expression of PPARa and FGF21 transcripts in vivo. Am. J. Physiol.—Endocrinol. Metab. 2010, 299, E607–E614. [Google Scholar]
- Arafat, A.M.; Kaczmarek, P.; Skrzypski, M.; Pruszynska-Oszmalek, E.; Kolodziejski, P.; Szczepankiewicz, D.; Sassek, M.; Wojciechowicz, T.; Wiedenmann, B.; Pfeiffer, A.F.H.; et al. Glucagon increases circulating fibroblast growth factor 21 independently of endogenous insulin levels: A novel mechanism of glucagon-stimulated lipolysis? Diabetologia 2013, 56, 588–597. [Google Scholar] [CrossRef]
- Lundåsen, T.; Hunt, M.C.; Nilsson, L.-M.; Sanyal, S.; Angelin, B.; Alexson, S.E.H.; Rudling, M. PPARa is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar]
- Pohjanvirta, R.; Sankari, S.; Kulju, T.; Naukkarinen, A.; Ylinen, M.; Tuomisto, J. Studies on the role of lipid peroxidation in the acute toxicity of TCDD in rats. Pharmacol. Toxicol. 1990, 66, 399–408. [Google Scholar] [CrossRef]
- Fletcher, N.; Wahlstrom, D.; Lundberg, R.; Nilsson, C.B.; Nilsson, K.C.; Stockling, K.; Hellmold, H.; Håkansson, H. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) alters the mRNA expression of critical genes associated with cholesterol metabolism, bile acid biosynthesis, and bile transport in rat liver: A microarray study. Toxicol. Appl. Pharmacol. 2005, 207, 1–24. [Google Scholar]
- Christian, B.J.; Menahan, L.A.; Peterson, R.E. Intermediary metabolism of the mature rat following 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment. Toxicol. Appl. Pharmacol. 1986, 83, 360–378. [Google Scholar] [CrossRef]
- Lakshman, M.R.; Ghosh, P.; Chirtel, S.J. Mechanism of action of 2,3,7,8-tetrachlorodibenzo-p-dioxin on intermediary metabolism in the rat. J. Pharmacol. Exp. Ther. 1991, 258, 317–319. [Google Scholar]
- Yao, C.Q.; Prokopec, S.D.; Watson, J.D.; Pang, R.; P’ng, C.; Chong, L.C.; Harding, N.J.; Pohjanvirta, R.; Okey, A.B.; Boutros, P.C. Inter-strain heterogeneity in rat hepatic transcriptomic responses to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol. Appl. Pharmacol. 2012, 260, 135–145. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Boutros, P.C.; Moffat, I.D.; Lindén, J.; Wendelin, D.; Okey, A.B. Genome-wide effects of acute progressive feed restriction in liver and white adipose tissue. Toxicol. Appl. Pharmacol. 2008, 230, 41–56. [Google Scholar] [CrossRef]
- Boone, L.; Meyer, D.; Cusick, P.; Ennulat, D.; Provencher Bolliger, A.; Everds, N.; Meador, V.; Elliott, G.; Honor, D.; Bounous, D.; et al. Selection and interpretation of clinical pathology indicators of hepatic injury in preclinical studies. Vet. Clin. Pathol. 2005, 34, 182–188. [Google Scholar] [CrossRef]
- Ennulat, D.; Magid-Slav, M.; Rehm, S.; Tatsuoka, K.S. Diagnostic performance of traditional hepatobiliary biomarkers of drug-induced liver injury in the rat. Toxicol. Sci. 2010, 116, 397–412. [Google Scholar] [CrossRef]
- Giannone, J.V.; Li, W.; Probst, M.; Okey, A.B. Prolonged depletion of AH receptor without alteration of receptor mRNA levels after treatment of cells in culture with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biochem. Pharmacol. 1998, 55, 489–497. [Google Scholar] [CrossRef]
- Pollenz, R.S. The mechanism of AH receptor protein down-regulation (degradation) and its impact on AH receptor-mediated gene regulation. Chem. Biol. Interact. 2002, 141, 41–61. [Google Scholar] [CrossRef]
- Franc, M.A.; Pohjanvirta, R.; Tuomisto, J.; Okey, A.B. In vivo up-regulation of aryl hydrocarbon receptor expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in a dioxin-resistant rat model. Biochem. Pharmacol. 2001, 62, 1565–1578. [Google Scholar] [CrossRef]
- MacPherson, L.; Ahmed, S.; Tamblyn, L.; Krutmann, J.; Förster, I.; Weighardt, H.; Matthews, J. Aryl hydrocarbon receptor repressor and TiPARP (ARTD14) use similar, but also distinct mechanisms to repress aryl hydrocarbon receptor signaling. Int. J. Mol. Sci. 2014, 15, 7939–7957. [Google Scholar] [CrossRef]
- Haarmann-Stemmann, T.; Abel, J. The arylhydrocarbon receptor repressor (AhRR): Structure, expression, and function. Biol. Chem. 2006, 387, 1195–1199. [Google Scholar]
- Hahn, M.E.; Allan, L.L.; Sherr, D.H. Regulation of constitutive and inducible AHR signaling: Complex interactions involving the AHR repressor. Biochem. Pharmacol. 2009, 77, 485–497. [Google Scholar] [CrossRef]
- Dietrich, C. The AHR in the control of cell cycle and apoptosis. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley and Sons: Hoboken, NJ, USA, 2012; pp. 467–484. [Google Scholar]
- Jackson, D.P.; Li, H.; Mitchell, K.A.; Joshi, A.D.; Elferink, C.J. Ah receptor-mediated suppression of liver regeneration through NC-XRE-driven p21Cip1 expression. Mol. Pharmacol. 2014, 85, 533–541. [Google Scholar] [CrossRef]
- Mitchell, K.A.; Lockhart, C.A.; Huang, G.; Elferink, C.J. Sustained aryl hydrocarbon receptor activity attenuates liver regeneration. Mol. Pharmacol. 2006, 70, 163–170. [Google Scholar]
- Lee, J.S.; Cella, M.; McDonald, K.G.; Garlanda, C.; Kennedy, G.D.; Nukaya, M.; Mantovani, A.; Kopan, R.; Bradfield, C.A.; Newberry, R.D.; et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2011, 13, 144–151. [Google Scholar] [CrossRef]
- Lahvis, G.P.; Bradfield, C.A. Ahr null alleles: Distinctive or different? Biochem. Pharmacol. 1998, 56, 781–787. [Google Scholar]
- Zhu, B.T.; Gallo, M.A.; Burger, C.W.; Meeker, R.J.; Cai, M.X.; Xu, S.; Conney, A.H. Effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin administration and high-fat diet on the body weight and hepatic estrogen metabolism in female C3H/HeN mice. Toxicol. Appl. Pharmacol. 2008, 226, 107–118. [Google Scholar] [CrossRef]
- Lindén, J.; Ranta, J.; Pohjanvirta, R. Bayesian modeling of reproducibility and robustness of RNA reverse transcription and quantitative real-time polymerase chain reaction. Anal. Biochem. 2012, 428, 81–91. [Google Scholar] [CrossRef]
- Bustin, S.A. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): Trends and problems. J. Mol. Endocrinol. 2002, 29, 23–39. [Google Scholar] [CrossRef]
- Tichopad, A.; Kitchen, R.; Riedmaier, I.; Becker, C.; Ståhlberg, A.; Kubista, M. Design and optimization of reverse-transcription quantitative PCR experiments. Clin. Chem. 2009, 55, 1816–1823. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lindén, J.; Lensu, S.; Pohjanvirta, R. Effect of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) on Hormones of Energy Balance in a TCDD-Sensitive and a TCDD-Resistant Rat Strain. Int. J. Mol. Sci. 2014, 15, 13938-13966. https://doi.org/10.3390/ijms150813938
Lindén J, Lensu S, Pohjanvirta R. Effect of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) on Hormones of Energy Balance in a TCDD-Sensitive and a TCDD-Resistant Rat Strain. International Journal of Molecular Sciences. 2014; 15(8):13938-13966. https://doi.org/10.3390/ijms150813938
Chicago/Turabian StyleLindén, Jere, Sanna Lensu, and Raimo Pohjanvirta. 2014. "Effect of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) on Hormones of Energy Balance in a TCDD-Sensitive and a TCDD-Resistant Rat Strain" International Journal of Molecular Sciences 15, no. 8: 13938-13966. https://doi.org/10.3390/ijms150813938