Mammalian Cytochrome P450-Dependent Metabolism of Polychlorinated Dibenzo-p-dioxins and Coplanar Polychlorinated Biphenyls

Abstract
:
1. Introduction


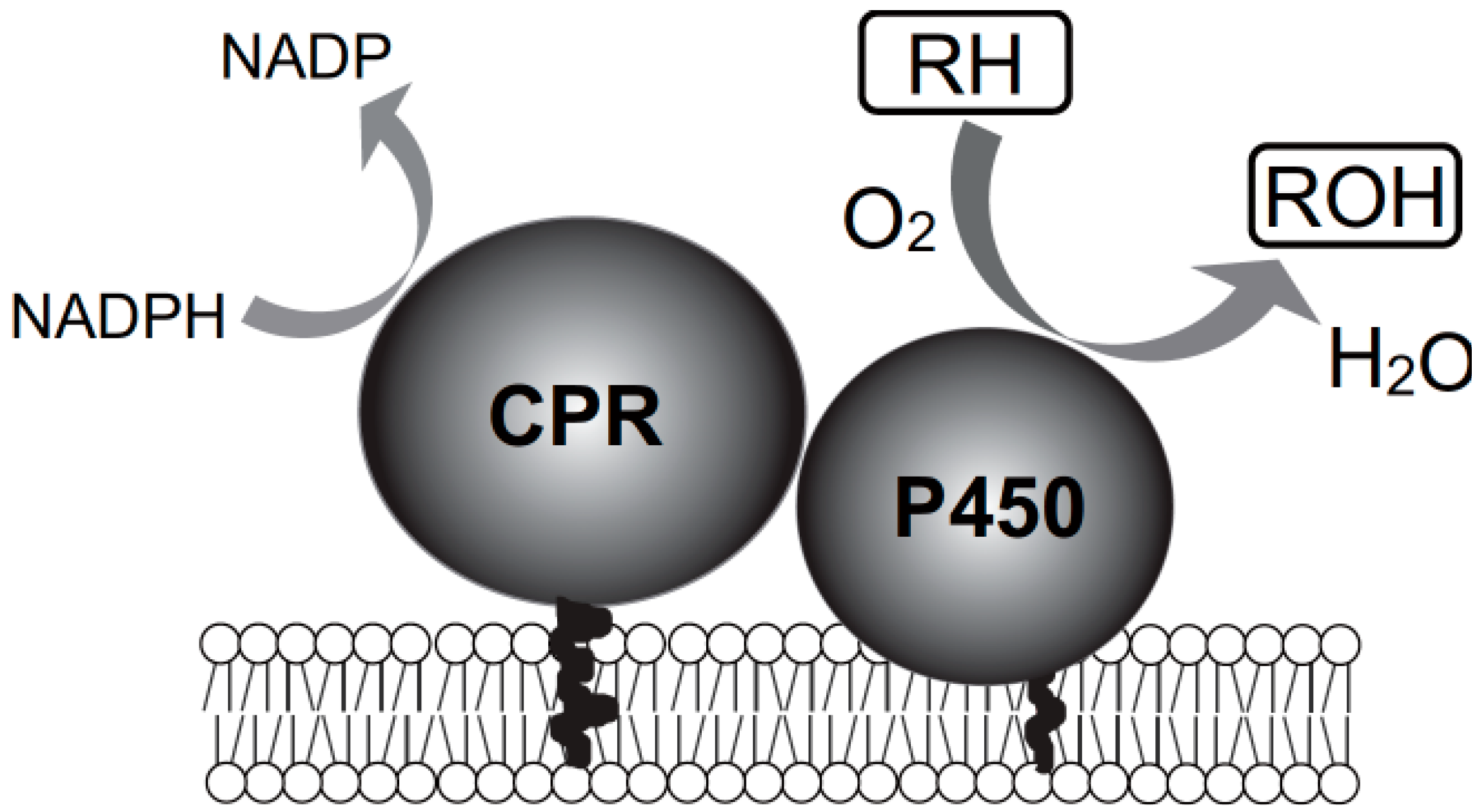
2. Yeast Expression System for Mammalian Cytochrome P450 (CYP) Isoforms
3. Metabolism of Polychlorinated Dibenzo-p-dioxins (PCDDs) by Human and Rat CYPs
3.1. Metabolism of 2,3,7,8-Tetrachlorodibenzo-p-dioxin (tetraCDD) in Humans

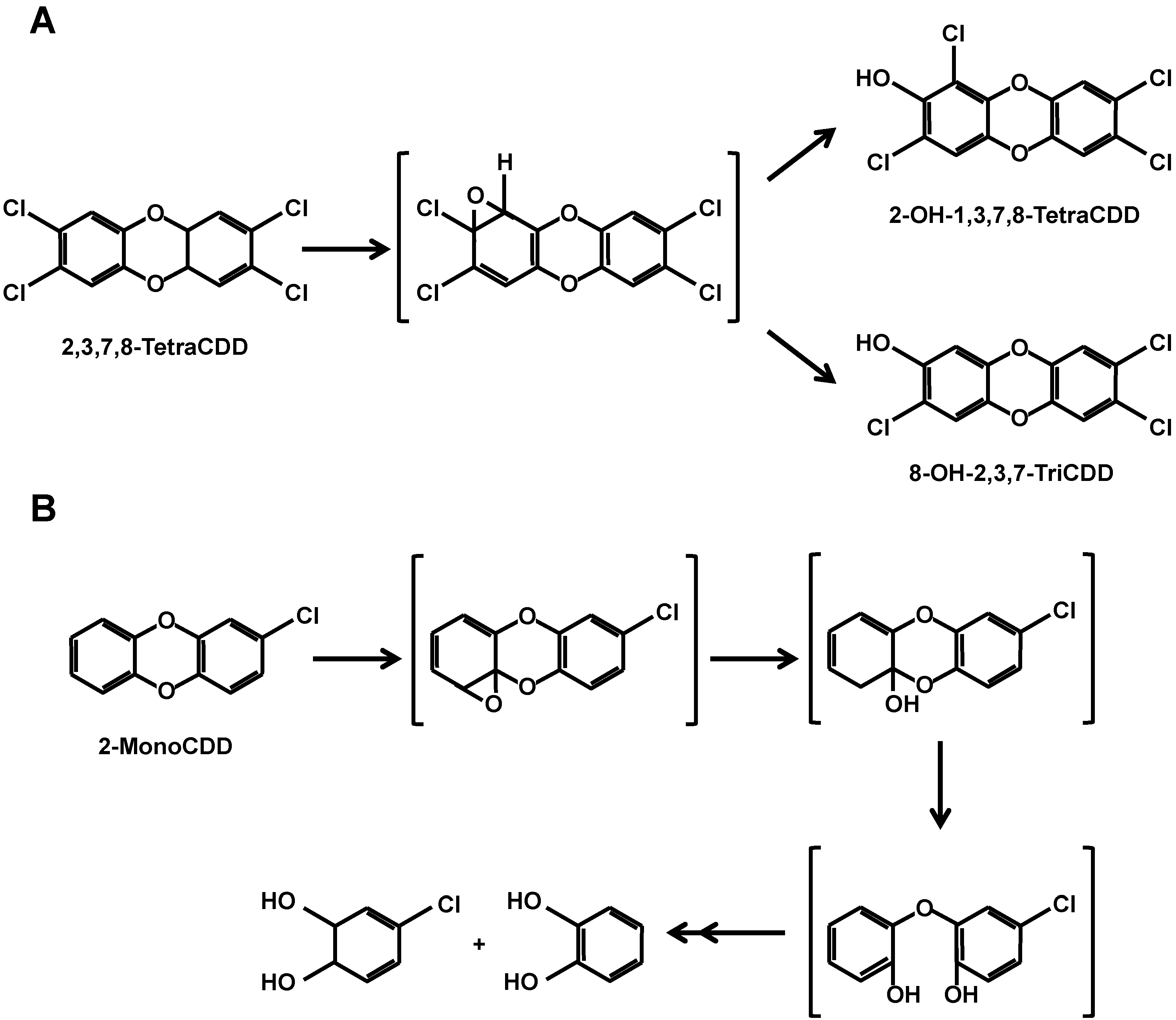
3.2. CYP-Dependent Metabolism of PCDDs
3.3. Binding of Tetra- and PentaCDDs to the Substrate-Binding Pocket of CYPs
3.4. Further Metabolism of PCDDs after CYP-Dependent Hydroxylation
3.5. Metabolism of 8-OH-2,3,7-triCDD by Human Recombinant UDP Glucuronosyltransferases (UGTs)
3.6. Successive Metabolism of 2,3,7-triCDD by CYPs and UGTs in Human Liver Microsomes
3.7. Species-Based Difference in CYP-Dependent Metabolism of PCDDs between Humans and Rats
3.8. Generation of 2,3,7,8-tetraCDD-Metabolizing CYPs by Modifying Rat CYP1A1 through Site-Directed Mutagenesis

4. Metabolism of PCBs by Human and Rat CYPs
4.1. In Vitro Metabolism of CB126 with Microsomal Fractions from Recombinant Yeast
4.2. Identification of CB126 Metabolites
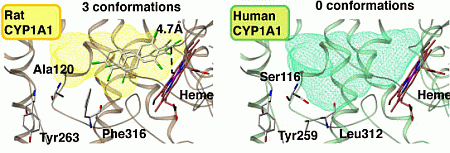
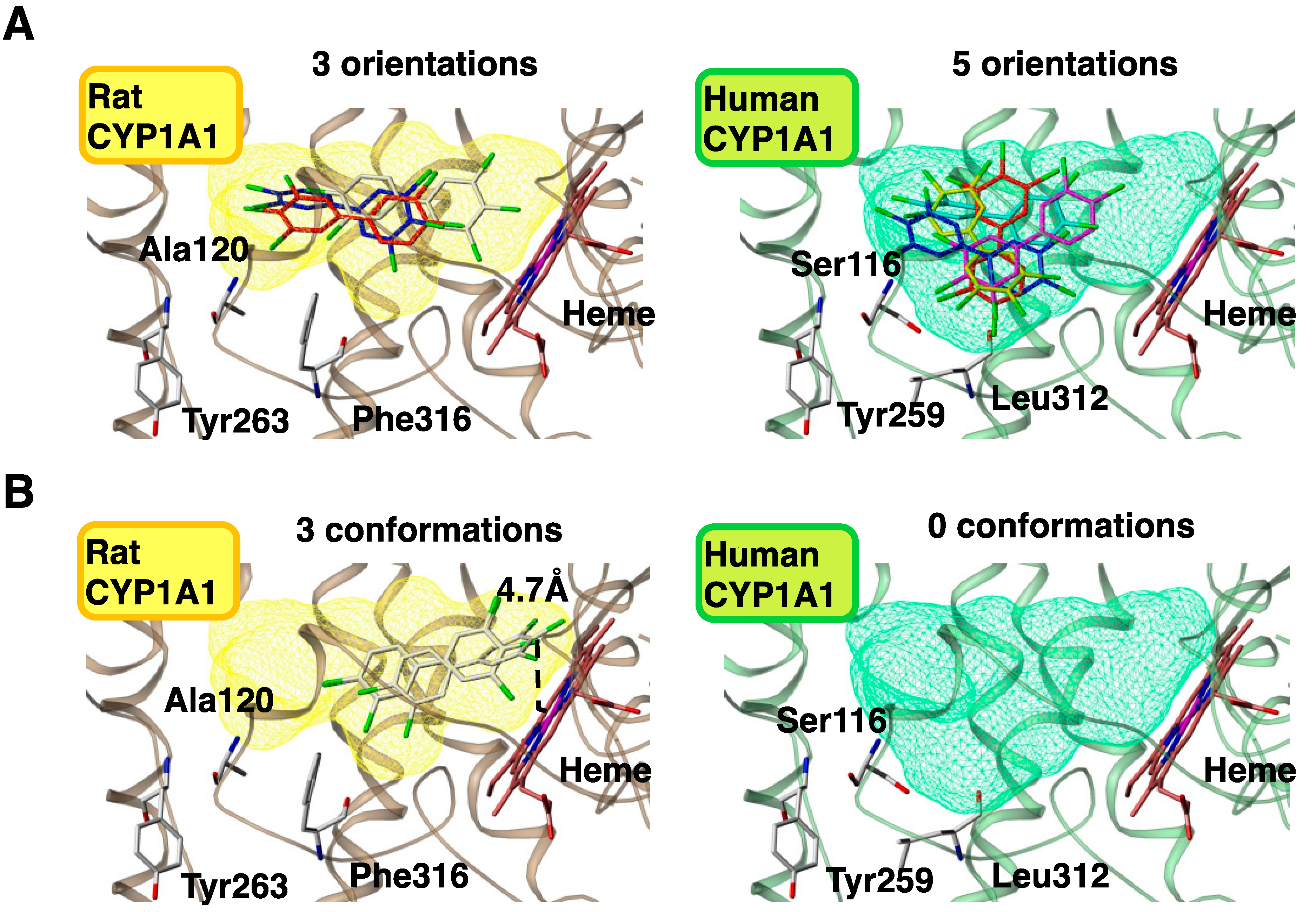
4.3. Molecular Modeling of Human and Rat CYP1A1s

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat | Human | Dog | Golden Hamster | Guinea Pig | Monkey | Mouse | Rabbit |
|---|---|---|---|---|---|---|---|
 |  |  |  |  |  |  |  |
| Ala | Ser | Thr | Thr | Ser | Ser | Thr | Thr |
 |  |  |  |  |  |  |  |
| Tyr | Tyr | Tyr | Tyr | Ser | His | Tyr | Tyr |
 |  |  |  |  |  |  |  |
| Phe | Leu | Leu | Val | Leu | Leu | Leu | Leu |

4.4. Construction of Docking Models with CB126 and CYP1A1s
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Leeuwen, F.X.; Feeley, M.; Schrenk, D.; Larsen, J.C.; Farland, W.; Younes, M. Dioxins: WHO’s tolerable daily intake (TDI) revisited. Chemosphere 2000, 40, 1095–1101. [Google Scholar] [CrossRef]
- Huwe, J.K. Dioxins in food: A modern agricultural perspective. J. Agric. Food Chem. 2002, 50, 1739–1750. [Google Scholar] [CrossRef]
- Ulaszewska, M.M.; Zuccato, E.; Davoli, E. PCDD/Fs and dioxin-like PCBs in human milk and estimation of infants’ daily intake: A review. Chemosphere 2011, 83, 774–782. [Google Scholar] [CrossRef]
- Rose, J.Q.; Ramsey, J.C.; Wentzler, T.H.; Hummel, R.A.; Gehring, P.J. The fate of 2,3,7,8-tetrachlorodibenzo-p-dioxin following single and repeated oral doses to the rat. Toxicol. Appl. Pharmacol. 1976, 36, 209–226. [Google Scholar] [CrossRef]
- Tulp, M.T.; Hutzinger, O. Identification of hydroxylated chlorodibenzo-p-dioxins, chlorodibenzofurans, chlorodiphenyl ethers and chloronaphthalenes as their methyl ethers by gas chromatography mass spectrometry. Biomed. Mass Spectrom. 1978, 5, 224–231. [Google Scholar] [CrossRef]
- Poiger, H.; Buser, H.R.; Weber, H.; Zweifel, U.; Schlatter, C. Structure elucidation of mammalian TCDD-metabolites. Experientia 1982, 38, 484–486. [Google Scholar] [CrossRef]
- Wroblewski, V.J.; Olson, J.R. Hepatic metabolism of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in the rat and guinea pig. Toxicol. Appl. Pharmacol. 1985, 81, 231–240. [Google Scholar] [CrossRef]
- Van den Berg, M.; de Jongh, J.; Poiger, H.; Olson, J.R. The toxicokinetics and metabolism of polychlorinated dibenzo-p-dioxins (PCDDs) and dibenzofurans (PCDFs) and their relevance for toxicity. Crit. Rev. Toxicol. 1994, 24, 1–74. [Google Scholar] [CrossRef]
- Hu, K.; Bunce, N.J. Metabolism of polychlorinated dibenzo-p-dioxins and related dioxin-like compounds. J. Toxicol. Environ. Health B Crit. Rev. 1999, 2, 183–210. [Google Scholar] [CrossRef]
- Huwe, J.K.; Feil, V.J.; Larsen, G.L.; Wiener, C. Metabolism and disposition of 1,4,7,8-tetrachlorodibenzo-p-dioxin in rats. Chemosphere 1998, 37, 1885–1893. [Google Scholar] [CrossRef]
- Sorg, O.; Zennegg, M.; Schmid, P.; Fedosyuk, R.; Valikhnovskyi, R.; Gaide, O.; Kniazevych, V.; Saurat, J.H. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) poisoning in Victor Yushchenko: Identification and measurement of TCDD metabolites. Lancet 2009, 374, 1179–1185. [Google Scholar] [CrossRef]
- Landi, M.T.; Bertazzi, P.A.; Baccarelli, A.; Consonni, D.; Masten, S.; Lucier, G.; Mocarelli, P.; Needham, L.; Caporaso, N.; Grassman, J. TCDD-mediated alterations in the AhR-dependent pathway in Seveso, Italy, 20 years after the accident. Carcinogenesis 2003, 24, 673–680. [Google Scholar] [CrossRef]
- Okino, S.T.; Quattrochi, L.C.; Pookot, D.; Iwahashi, M.; Dahiya, R. A dioxin-responsive enhancer 3' of the human CYP1A2 gene. Mol. Pharmacol. 2007, 72, 1457–1465. [Google Scholar] [CrossRef]
- Van den Berg, M.; Birnbaum, L.S.; Denison, M.; de Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization reevaluation of human and mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef]
- Wahlang, B.; Falkner, K.C.; Clair, H.B.; Al-Eryani, L.; Prough, R.A.; States, J.C.; Coslo, D.M.; Omiecinski, C.J.; Cave, M.C. Human receptor activation by Aroclor 1260, a polychlorinated biphenyl mixture. Toxicol. Sci. 2014. [Google Scholar] [CrossRef]
- Kishida, M.; Imamura, K.; Takenaka, N.; Maeda, Y.; Viet, P.H.; Kondo, A.; Bandow, H. Characteristics of the abundance of polychlorinated dibenzo-p-dioxin and dibenzofurans, and dioxin-like polychlorinated biphenyls in sediment samples from selected Asian regions in Can Gio, Southern Vietnam and Osaka, Japan. Chemosphere 2010, 78, 127–133. [Google Scholar] [CrossRef]
- Verreault, J.; Norstrom, R.J.; Ramsay, M.A.; Mulvihill, M.; Letcher, R.J. Composition of chlorinated hydrocarbon contaminants among major adipose tissue depots of polar bears (Ursus maritimus) from the Canadian high Arctic. Sci. Total Environ. 2006, 370, 580–587. [Google Scholar] [CrossRef]
- Koga, N.; Beppu, M.; Yoshimura, H. Metabolism in vivo of 3,4,5,3',4'-pentachlorobiphenyl and toxicological assessment of the metabolite in rats. J. Pharmacobiodyn. 1990, 13, 497–506. [Google Scholar] [CrossRef]
- Haraguchi, K.; Hirose, Y.; Masuda, Y.; Kato, Y.; Kimura, R. Metabolism of 3,3',4,4',5-penta- and 2,2',3,3',4,4'-hexachlorobiphenyls in rats. Fukuoka Igaku Zasshi 1999, 90, 210–219. [Google Scholar]
- Yamazaki, K.; Suzuki, M.; Itoh, T.; Yamamoto, K.; Kanemitsu, M.; Matsumura, C.; Nakano, T.; Sakaki, T.; Fukami, Y.; Imaishi, H.; et al. Structural basis of species differences between human and experimental animal CYP1A1s in metabolism of 3,3',4,4',5-pentachlorobiphenyl. J. Biochem. 2011, 149, 487–494. [Google Scholar] [CrossRef]
- Ohta, C.; Haraguchi, K.; Kato, Y.; Koga, N. In vitro metabolism of 2,2',3,4',5,5',6-heptachlorobiphenyl (CB187) by liver microsomes from rats, hamsters and guinea pigs. Xenobiotica 2005, 35, 319–330. [Google Scholar] [CrossRef]
- Oeda, K.; Sakaki, T.; Ohkawa, H. Expression of rat liver cytochrome P-450MC cDNA in Saccharomyces cerevisiae. DNA 1985, 4, 203–210. [Google Scholar] [CrossRef]
- Sakaki, T.; Oeda, K.; Yabusaki, Y.; Ohkawa, H. Monooxygenase activity of Saccharomyces cerevisiae cells transformed with expression plasmids carrying rat cytochrome P-450MC cDNA. J. Biochem. 1986, 99, 741–749. [Google Scholar]
- Bussey, H.; Storms, R.K.; Ahmed, A.; Albermann, K.; Allen, E.; Ansorge, W.; Araujo, R.; Aparicio, A.; Barrell, B.; Badcock, K.; et al. The nucleotide sequence of Saccharomyces cerevisiae chromosome XVI. Nature 1997, 387, 103–105. [Google Scholar]
- Sakaki, T.; Inouye, K. Practical application of mammalian cytochrome P450. J. Biosci. Bioeng. 2000, 90, 583–590. [Google Scholar]
- Imaoka, S.; Yamada, T.; Hiroi, T.; Hayashi, K.; Sakaki, T.; Yabusaki, Y.; Funae, Y. Multiple forms of human P450 expressed in Saccharomyces cerevisiae: Systematic characterization and comparison with those of the rat. Biochem. Pharmacol. 1996, 51, 1041–1050. [Google Scholar] [CrossRef]
- Hayashi, K.; Sakaki, T.; Kominami, S.; Inouye, K.; Yabusaki, Y. Coexpression of genetically engineered fused enzyme between yeast NADPH–P450 reductase and human cytochrome P450 3A4 and human cytochrome b5 in yeast. Arch. Biochem. Biophys. 2000, 381, 164–170. [Google Scholar] [CrossRef]
- Sakaki, T.; Munetsuna, E. Enzyme systems for biodegradation of polychlorinated dibenzo-p-dioxins. Appl. Microbiol. Biotechnol. 2010, 88, 23–30. [Google Scholar] [CrossRef]
- Inouye, K.; Shinkyo, R.; Takita, T.; Ohta, M.; Sakaki, T. Metabolism of polychlorinated dibenzo-p-dioxins (PCDDs) by human cytochrome P450-dependent monooxygenase systems. J. Agric. Food Chem. 2002, 50, 5496–5502. [Google Scholar] [CrossRef]
- Staskal, D.F.; Diliberto, J.J.; Devito, M.J.; Birnbaum, L.S. Inhibition of human and rat CYP1A2 by TCDD and dioxin-like chemicals. Toxicol. Sci. 2005, 84, 225–231. [Google Scholar] [CrossRef]
- Hakk, H.; Larsen, G.; Feil, V. Tissue distribution, excretion, and metabolism of 1,2,7,8-tetrachlorodibenzo-p-dioxin in the rat. Chemosphere 2001, 42, 975–983. [Google Scholar] [CrossRef]
- Mackenzie, P.I.; Bock, K.W.; Burchell, B.; Guillemette, C.; Ikushiro, S.; Iyanagi, T.; Miners, J.O.; Owens, I.S.; Nebert, D.W. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet. Genomics 2005, 15, 677–685. [Google Scholar] [CrossRef]
- Kasai, N.; Sakaki, T.; Shinkyo, R.; Ikushiro, S.; Iyanagi, T.; Kamao, M.; Okano, T.; Ohta, M.; Inouye, K. Sequential metabolism of 2,3,7-trichlorodibenzo-p-dioxin (2,3,7-triCDD) by cytochrome P450 and UDP-glucuronosyltransferase in human liver microsomes. Drug Metab. Dispos. 2004, 32, 870–875. [Google Scholar] [CrossRef]
- Mason, G.; Safe, S. Synthesis, biologic and toxic effects of the major 2,3,7,8-tetrachlorodibenzo-p-dioxin metabolites in the rat. Toxicology 1986, 41, 153–159. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar]
- Von Moltke, L.L.; Greenblatt, D.J.; Duan, S.X.; Schmider, J.; Kudchadker, L.; Fogelman, S.M.; Harmatz, J.S.; Shader, R.I. Phenacetin O-deethylation by human liver microsomes in vitro: Inhibition by chemical probes, SSRI antidepressants, nefazodone and venlafaxine. Psychopharmacology 1996, 128, 398–407. [Google Scholar] [CrossRef]
- Shinkyo, R.; Sakaki, T.; Ohta, M.; Inouye, K. Metabolic pathways of dioxin by CYP1A1: Species difference between rat and human CYP1A subfamily in the metabolism of dioxins. Arch. Biochem. Biophys. 2003, 409, 180–187. [Google Scholar] [CrossRef]
- Shinkyo, R.; Sakaki, T.; Takita, T.; Ohta, M.; Inouye, K. Generation of 2,3,7,8-TCDD-metabolizing enzyme by modifying rat CYP1A1 through site-directed mutagenesis. Biochem. Biophys. Res. Commun. 2003, 308, 511–517. [Google Scholar] [CrossRef]
- Sansen, S.; Yano, J.K.; Reynald, R.L.; Schoch, G.A.; Griffin, K.J.; Stout, C.D.; Johnson, E.F. Adaptations for the oxidation of polycyclic aromatic hydrocarbons exhibited by the structure of human P450 1A2. J. Biol. Chem. 2007, 282, 14348–14355. [Google Scholar]
- Itoh, T.; Takemura, H.; Shimoi, K.; Yamamoto, K. A 3D model of CYP1B1 explains the dominant 4-hydroxylation of estradiol. J. Chem. Inf. Model. 2010, 50, 1173–1178. [Google Scholar] [CrossRef]
- Sakiyama, T.; Yamamoto, A.; Kakutani, N.; Fukuyama, J.; Okumura, T. Hydroxylated polychlorinated biphenyls (OH–PCBs) in the aquatic environment: Levels and congener profiles in sediments from Osaka, Japan. Organohalogen Comp. 2007, 71, 1881–1885. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Inui, H.; Itoh, T.; Yamamoto, K.; Ikushiro, S.-I.; Sakaki, T. Mammalian Cytochrome P450-Dependent Metabolism of Polychlorinated Dibenzo-p-dioxins and Coplanar Polychlorinated Biphenyls. Int. J. Mol. Sci. 2014, 15, 14044-14057. https://doi.org/10.3390/ijms150814044
Inui H, Itoh T, Yamamoto K, Ikushiro S-I, Sakaki T. Mammalian Cytochrome P450-Dependent Metabolism of Polychlorinated Dibenzo-p-dioxins and Coplanar Polychlorinated Biphenyls. International Journal of Molecular Sciences. 2014; 15(8):14044-14057. https://doi.org/10.3390/ijms150814044
Chicago/Turabian StyleInui, Hideyuki, Toshimasa Itoh, Keiko Yamamoto, Shin-Ichi Ikushiro, and Toshiyuki Sakaki. 2014. "Mammalian Cytochrome P450-Dependent Metabolism of Polychlorinated Dibenzo-p-dioxins and Coplanar Polychlorinated Biphenyls" International Journal of Molecular Sciences 15, no. 8: 14044-14057. https://doi.org/10.3390/ijms150814044