Identification of Diversity-Generating Retroelements in Human Microbiomes

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Testing of DGRscan on Known DGR Systems

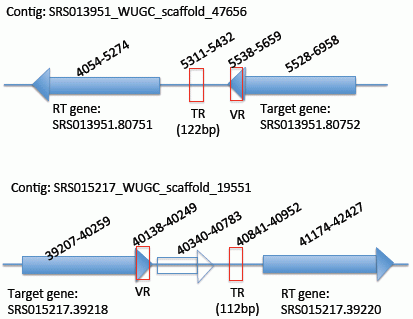
{kind=link}
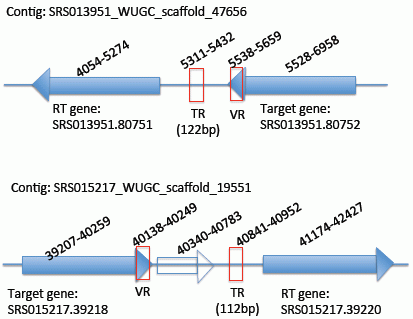
{kind=link}
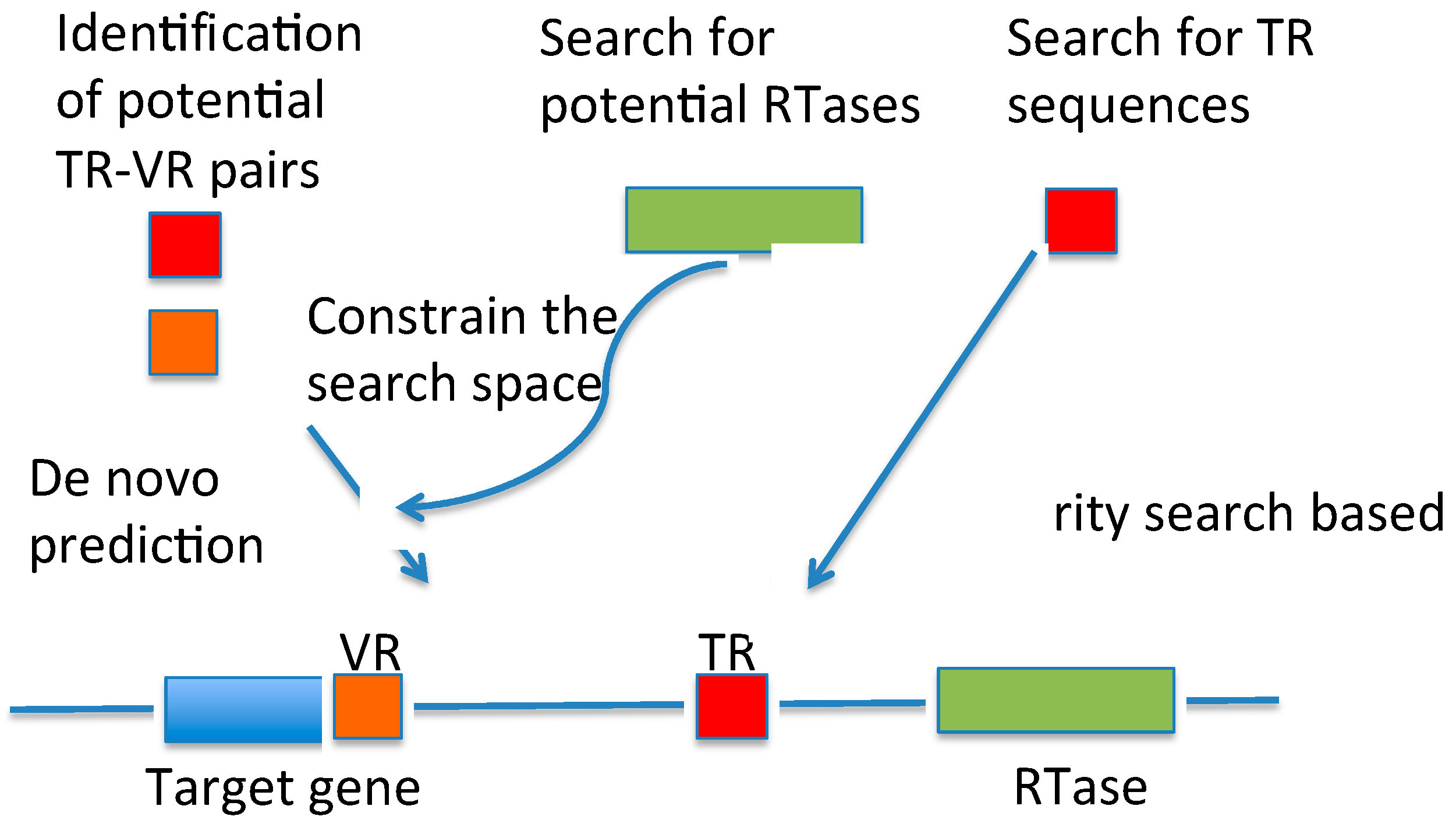
{kind=link}
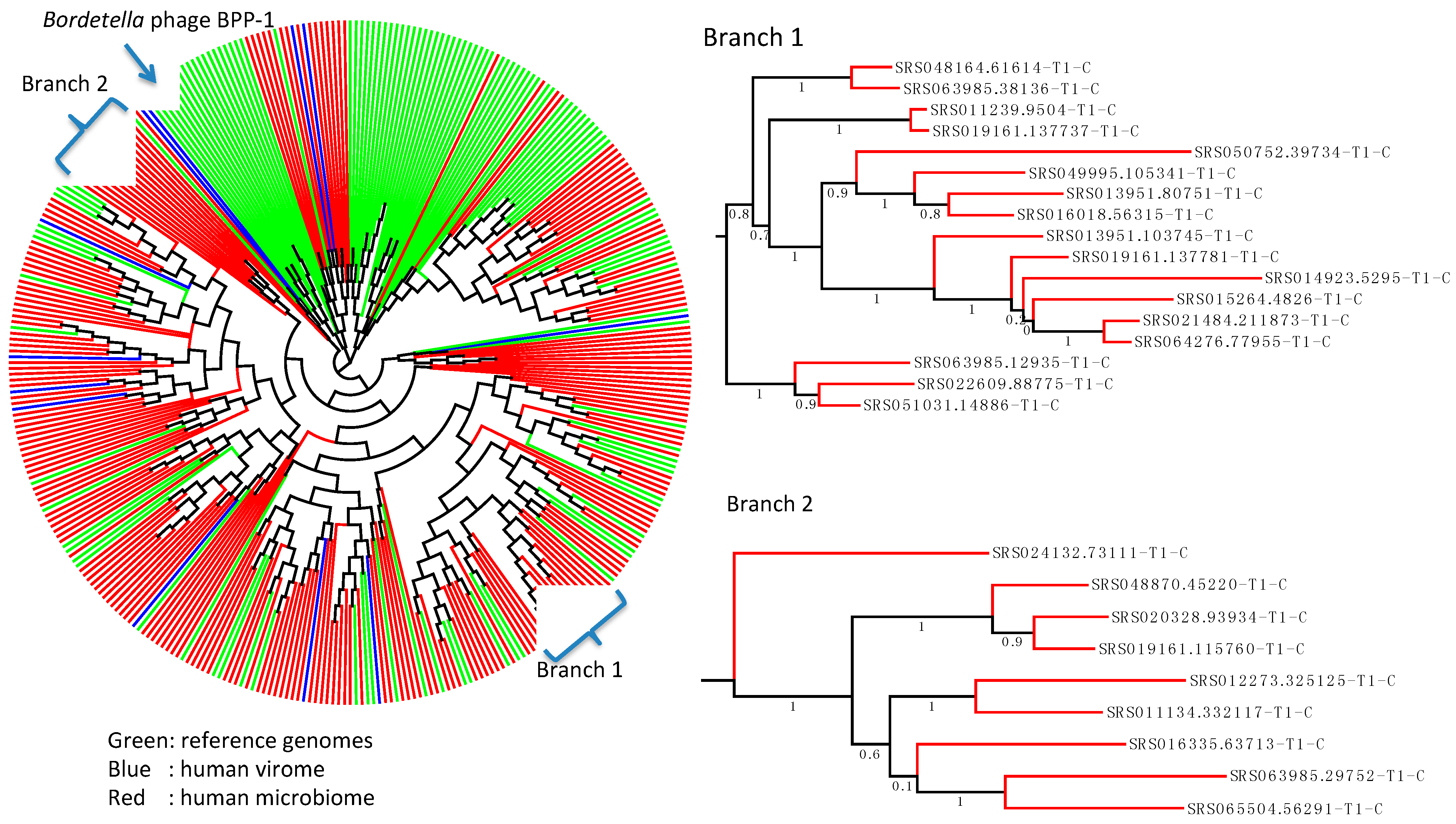
{kind=link}
{kind=link}
| Sequence (gi) | Previous Study [7] | DGRscan | ||
|---|---|---|---|---|
| Length (bp) | Start–End | Length (bp) | Start–End | |
| gi|377805826|gb|JQ680351.1| | 101 | 22,583–22,683 | 112 | 22,869–22,980 |
| gi|377805855|gb|JQ680352.1| | 139 | 7998–8136 | 131 | 7998–8128 |
| gi|377805862|gb|JQ680353.1| | 89 | 14,041–14,129 | 128 | 14,041–14,168 |
| gi|377805758|gb|JQ680349.1| | 97 | 39,459–39,555 | 126 | 39,449–39,574 |
| gi|377805877|gb|JQ680354.1| | 128 | 46,739–46,866 | 128 | 46,739–46,866 |
| gi|377805785|gb|JQ680350.1| | 101 | 17,450–17,550 | 128 | 17,450–17,577 |
| gi|377805936|gb|JQ680355.1| | 101 | 3782–3882 | 94 | 3782–3875 |
| gi|377806015|gb|JQ680359.1| | 116 | 4435–4550 | 126 | 4435–4560 |
| gi|377805967|gb|JQ680357.1| | 101 | 3148–3248 | 158 | 3091–3248 |
| gi|377806003|gb|JQ680358.1| | 128 | 5598–5725 | 128 | 5598–5725 |
| gi|377805941|gb|JQ680356.1| | 128 | 38,849–38,976 | 128 | 38,849–38,976 |
| gi|377806018|gb|JQ680360.1| | 23 | 6556–6578 | 116 | 6463–6578 |
| gi|377806060|gb|JQ680361.1| | 78 | 21,703–21,780 | 116 | 21,692–21,807 |
| gi|377806090|gb|JQ680362.1| | 121 | 4920–5040 | 122 | 4918–5039 |
| gi|377806097|gb|JQ680363.1| | 101 | 4306–4406 | 110 | 4306–4415 |
| gi|377806107|gb|JQ680364.1| | 121 | 11,625–11,745 | 122 | 11,620–11,741 |
| gi|377806133|gb|JQ680365.1| | 101 | 32,701–32,801 | 94 | 32,685–32,778 |
| gi|377806170|gb|JQ680366.1| | 101 | 8872–8972 | 113 | 8818–8930 |
| gi|377806186|gb|JQ680367.1| | 120 | 41,651–41,770 | 123 | 42,437–42,559 |
| gi|377806226|gb|JQ680368.1| | 121 | 27,017–27,137 | 126 | 27,012–27,137 |
| gi|377806251|gb|JQ680369.1| | 85 | 3464–3548 | 115 | 3464–3578 |
| gi|377806260|gb|JQ680370.1| | 115 | 25,676–25,790 | 115 | 25,676–25,790 |
| gi|377806297|gb|JQ680372.1| | 151 | 2108–2258 | 121 | 2108–2228 |
| gi|377806292|gb|JQ680371.1| | 59 | 4471–4529 | 110 | 4471–4580 |
| gi|377806399|gb|JQ680376.1| | 44 | 3993–4036 | - | - |
| gi|377806374|gb|JQ680375.1| | 101 | 23,576–23,676 | 75 | 23,569–23,643 |
| gi|377806345|gb|JQ680374.1| | 101 | 4412–4512 | 95 | 4420–4514 |
| gi|377806301|gb|JQ680373.1| | 101 | 20,259–20,359 | 133 | 20,252–20,384 |
| gi|377806422|gb|JQ680377.1| | 121 | 13,118–13,238 | 105 | 13,134–13,238 |
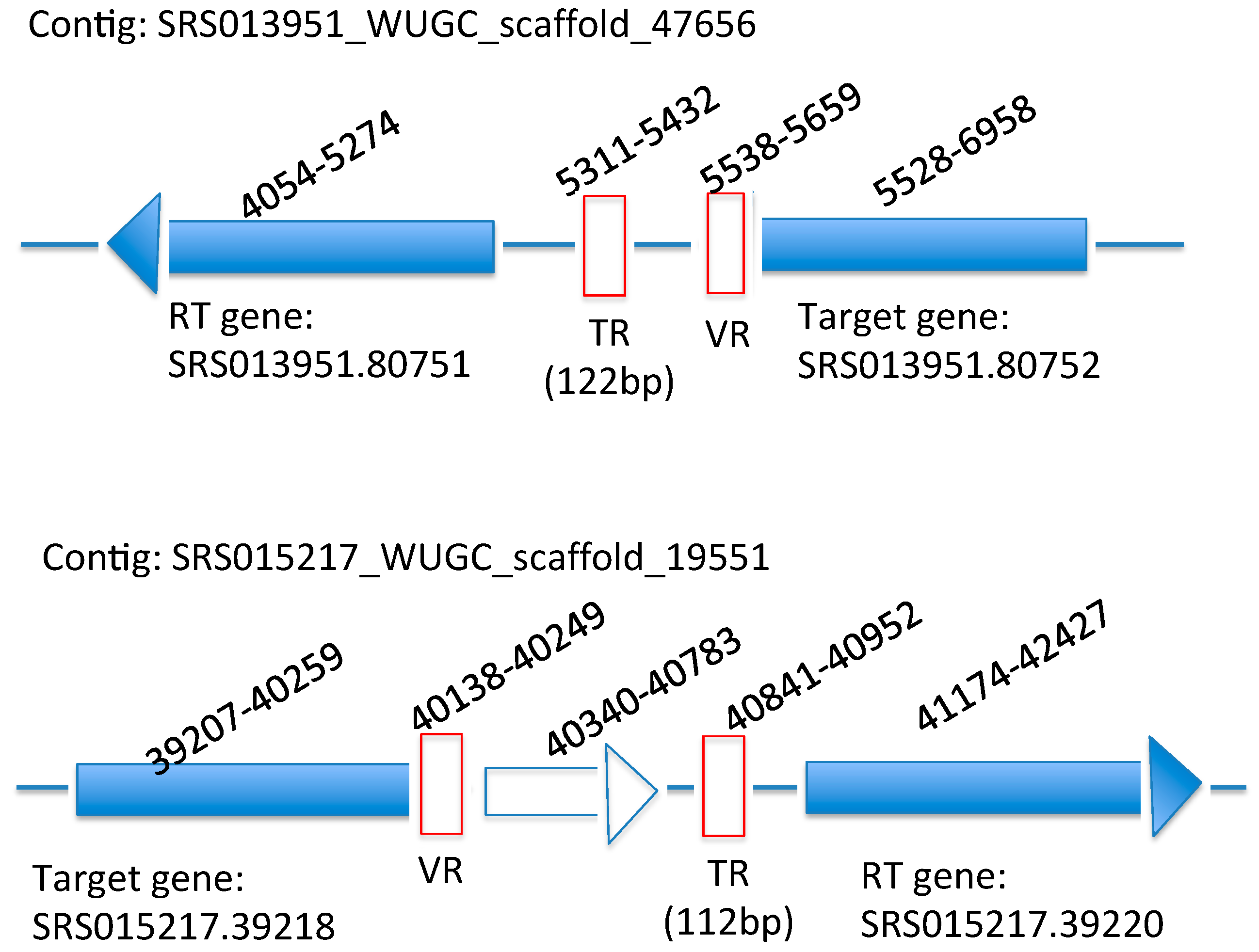
2.2. Identification of the DGR Systems Associated with the Human Microbiome

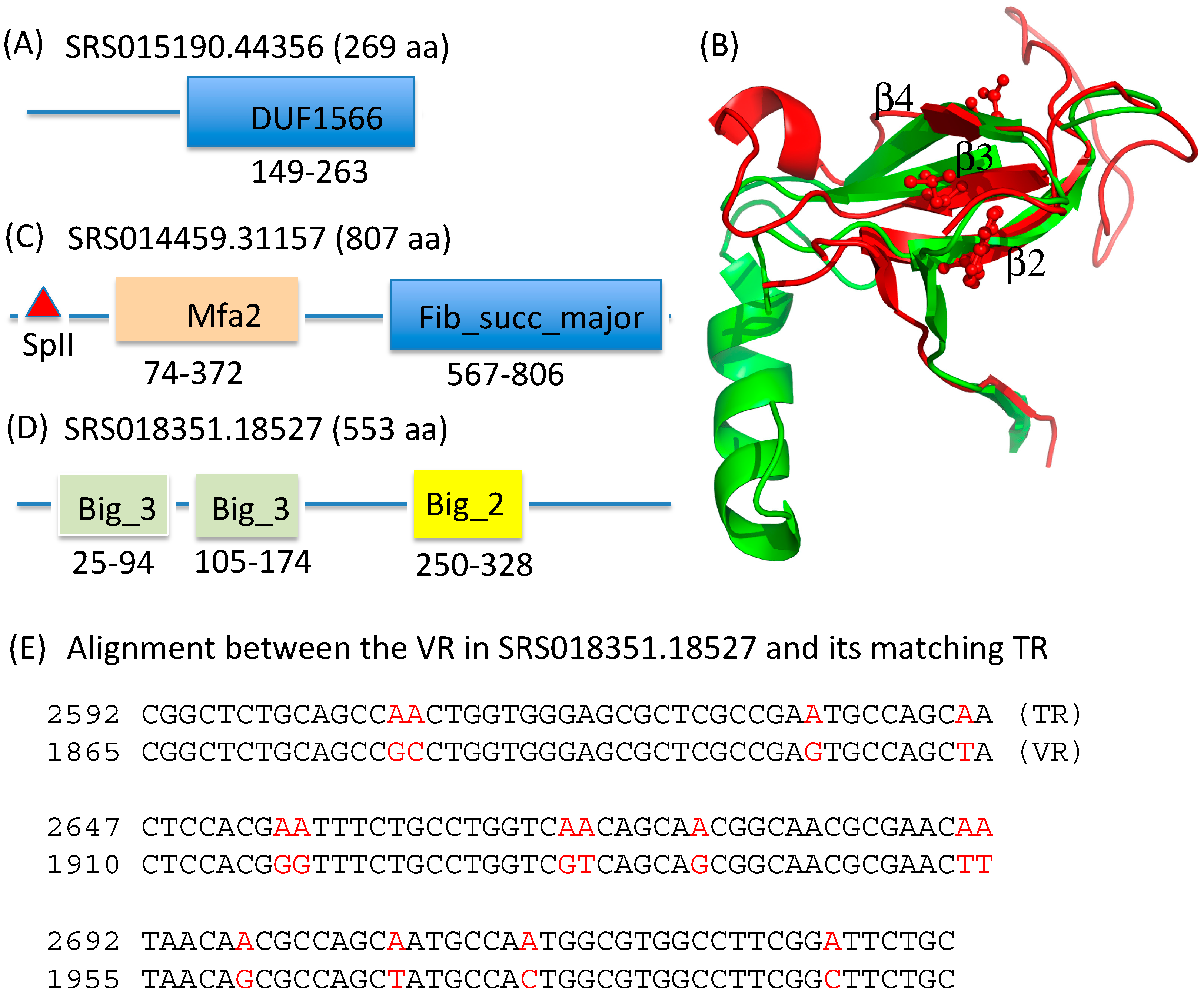
2.3. Identification and Annotation of the Target Genes

| Pfam Domain | Description | Number of Proteins | Example |
|---|---|---|---|
| DUF1566 | Protein of unknown function; similar to Fib_succ_major | 47 | SRS015190.44356 |
| FGE-sulfatase | Sulfatase-modifying factor enzyme 1 | 42 | SRS052027.8709 |
| Fib_succ_major | Fibrobacter succinogenes major domain | 9 | SRS014459.31157 |
| DUF3988 | Found by clustering human gut metagenomic sequences | 9 | SRS077730.37228 |
| Big_2 | Bacterial Ig-like domain (group 2) | 9 | SRS018351.18527 |
| DUF3751 | Phage tail-collar fiber protein | 8 | SRS013687.79027 |
| CotH | Members of this family include the spore coat protein H | 6 | SRS016989.185 |
| Big_3 | Bacterial Ig-like domain (group 3) | 6 | SRS018351.18527 |
| CarboxypepD_reg | Carboxypeptidase regulatory-like domain | 5 | SRS015663.100342 |
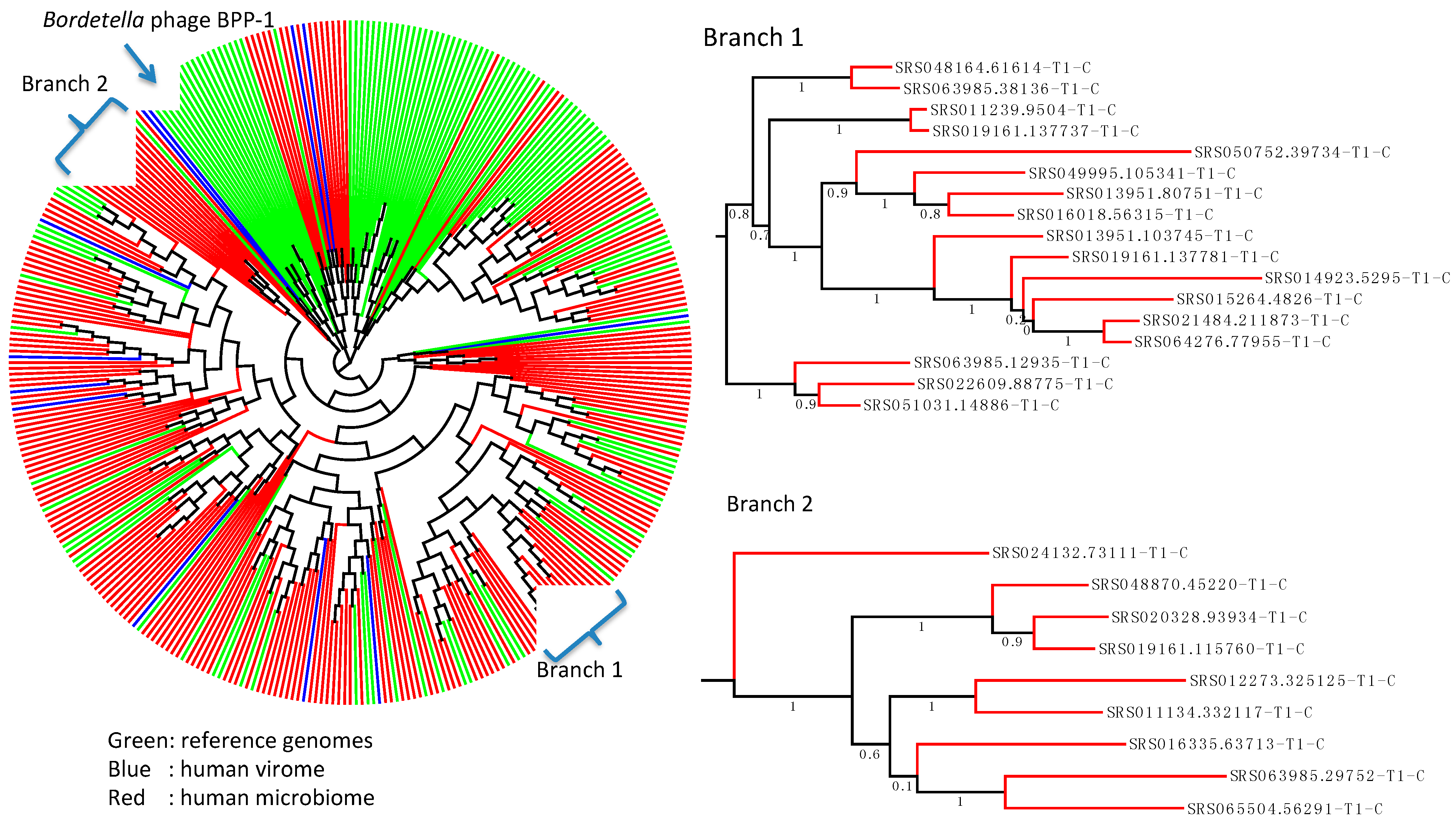
2.4. Wide Distribution of the DGR Systems in the Human Microbiome
3. Experimental Section
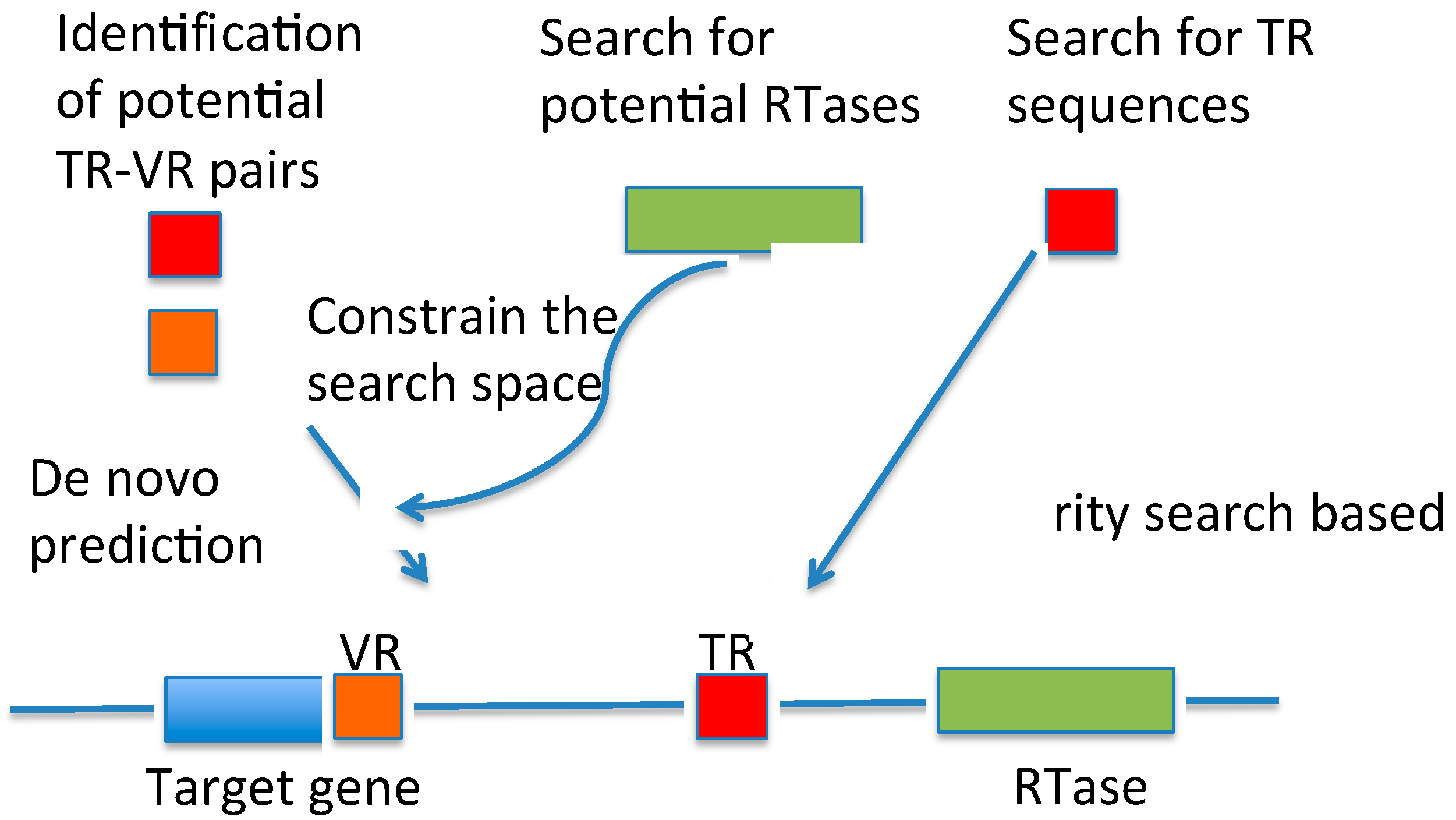
3.1. DGRscan
3.2. The Human Virome Datasets and the Human Microbiome Project (HMP) Datasets
3.3. Availability of DGRscan and Results
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Doulatov, S.; Hodes, A.; Dai, L.; Mandhana, N.; Liu, M.; Deora, R.; Simons, R.W.; Zimmerly, S.; Miller, J.F. Tropism switching in Bordetella bacteriophage defines a family of diversity-generating retroelements. Nature 2004, 431, 476–481. [Google Scholar] [CrossRef]
- Guo, H.; Tse, L.V.; Nieh, A.W.; Czornyj, E.; Williams, S.; Oukil, S.; Liu, V.B.; Miller, J.F. Target site recognition by a diversity-generating retroelement. PLoS Genet. 2011, 7, e1002414. [Google Scholar] [CrossRef]
- Alayyoubi, M.; Guo, H.; Dey, S.; Golnazarian, T.; Brooks, G.A.; Rong, A.; Miller, J.F.; Ghosh, P. Structure of the essential diversity-generating retroelement protein bAvd and its functionally important interaction with reverse transcriptase. Structure 2013, 21, 266–276. [Google Scholar] [CrossRef]
- Medhekar, B.; Miller, J.F. Diversity-generating retroelements. Curr. Opin. Microbiol. 2007, 10, 388–395. [Google Scholar] [CrossRef]
- Schillinger, T.; Zingler, N. The low incidence of diversity-generating retroelements in sequenced genomes. Mob. Genet. Elem. 2012, 2, 287–291. [Google Scholar] [CrossRef]
- Le Coq, J.; Ghosh, P. Conservation of the C-type lectin fold for massive sequence variation in a Treponema diversity-generating retroelement. Proc. Natl. Acad. Sci. USA 2011, 108, 14649–14653. [Google Scholar] [CrossRef]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Arambula, D.; Wong, W.; Medhekar, B.A.; Guo, H.; Gingery, M.; Czornyj, E.; Liu, M.; Dey, S.; Ghosh, P.; Miller, J.F. Surface display of a massively variable lipoprotein by a Legionella diversity-generating retroelement. Proc. Natl. Acad. Sci. USA 2013, 110, 8212–8217. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [CrossRef] [Green Version]
- The Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [CrossRef] [Green Version]
- Schillinger, T.; Lisfi, M.; Chi, J.; Cullum, J.; Zingler, N. Analysis of a comprehensive dataset of diversity generating retroelements generated by the program DiGReF. BMC Genomics 2012, 13, 430. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Howard, M.T.; Satoshi, M. Viral RNA-dependent DNA polymerase: RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 1970, 226, 1211–1213. [Google Scholar] [CrossRef]
- Pfam RVT_1 Domain. Available online: http://pfam.xfam.org/family/PF00078 (accessed on 14 June 2014).
- Minot, S.; Grunberg, S.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Hypervariable loci in the human gut virome. Proc. Natl. Acad. Sci. USA 2012, 109, 3962–3966. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Han, M.V.; Zmasek, C.M. PhyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinform. 2009, 10, 356. [Google Scholar] [CrossRef]
- McMahon, S.A.; Miller, J.L.; Lawton, J.A.; Kerkow, D.E.; Hodes, A.; Marti-Renom, M.A.; Doulatov, S.; Narayanan, E.; Sali, A.; Miller, J.F.; et al. The C-type lectin fold as an evolutionary solution for massive sequence variation. Nat. Struct. Mol. Biol. 2005, 12, 886–892. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- The HMMER Server. Available online: http://hmmer.janelia.org/search/hmmscan (accessed on 12 June 2014).
- Pfam DUF1566. Available online: http://pfam.xfam.org/family/Duf1566 (accessed on 14 June 2014).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Kallberg, M.; Margaryan, G.; Wang, S.; Ma, J.; Xu, J. RaptorX server: A resource for template-based protein structure modeling. Methods Mol. Biol. 2014, 1137, 17–27. [Google Scholar] [CrossRef]
- The RaptorX Server. Available online: http://raptorx.uchicago.edu/ (accessed on 12 June 2014).
- Ye, Y.; Godzik, A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 2003, 19, II246–II255. [Google Scholar]
- Juncker, A.S.; Willenbrock, H.; von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef]
- DACC Website. Available online: http://www.hmpdacc.org/ (accessed on 20 March 2013).
- DGRscan Website. Available online: http://omics.informatics.indiana.edu/mg/DGRscan (accessed on 15 June 2014).
- DGRscan at Github. Available online: https://github.com/YuzhenYe/DGRscan (accessed on 15 June 2014).
- Zhang, Q.; Doak, T.G.; Ye, Y. Expanding the catalog of cas genes with metagenomes. Nucleic Acids Res. 2014, 42, 2448–2459. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ye, Y. Identification of Diversity-Generating Retroelements in Human Microbiomes. Int. J. Mol. Sci. 2014, 15, 14234-14246. https://doi.org/10.3390/ijms150814234
Ye Y. Identification of Diversity-Generating Retroelements in Human Microbiomes. International Journal of Molecular Sciences. 2014; 15(8):14234-14246. https://doi.org/10.3390/ijms150814234
Chicago/Turabian StyleYe, Yuzhen. 2014. "Identification of Diversity-Generating Retroelements in Human Microbiomes" International Journal of Molecular Sciences 15, no. 8: 14234-14246. https://doi.org/10.3390/ijms150814234