Cross-Species, Amplifiable Microsatellite Markers for Neoverrucid Barnacles from Deep-Sea Hydrothermal Vents Developed Using Next-Generation Sequencing

Abstract
:1. Introduction
2. Results and Discussion

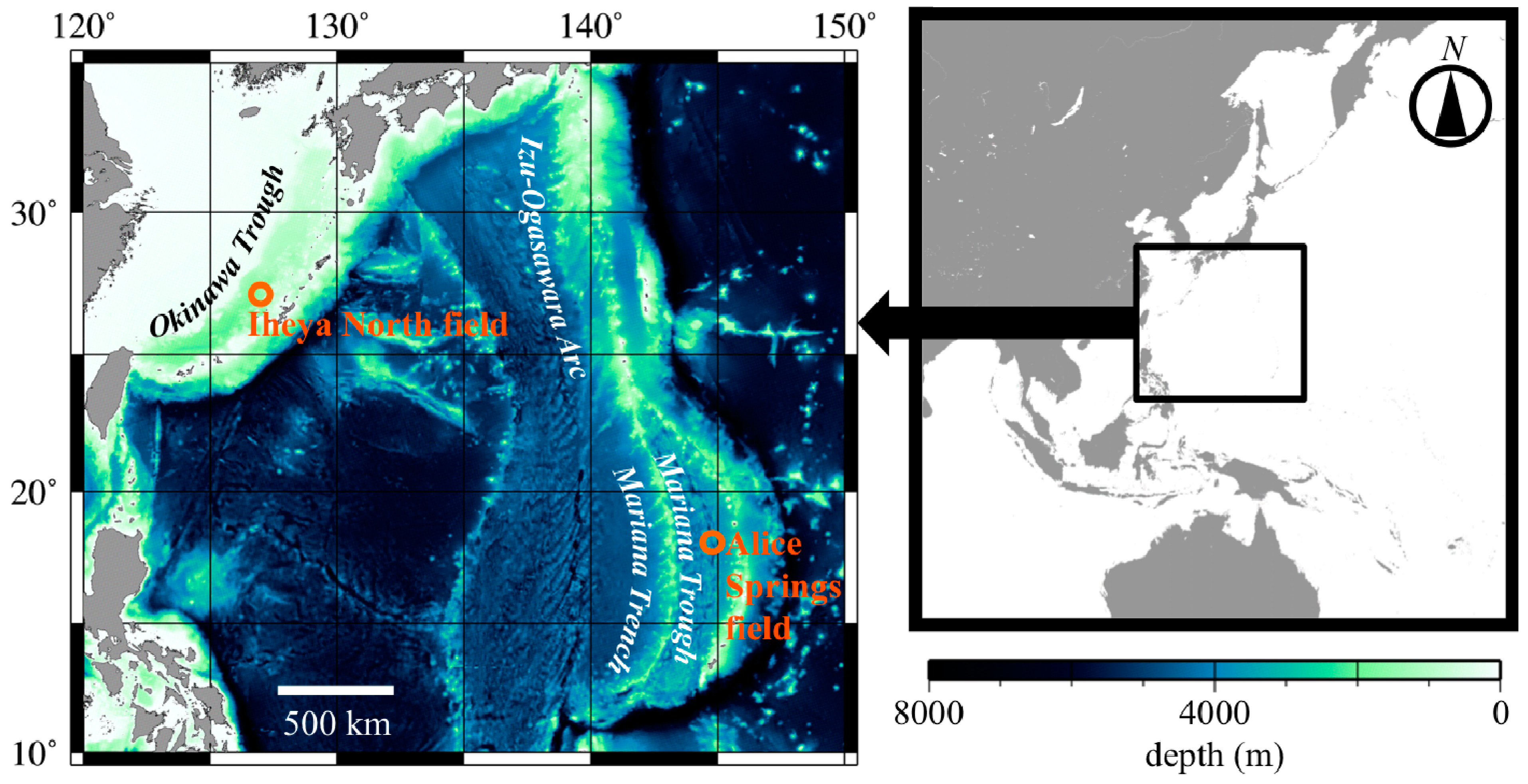{kind=link}
| Locus | Repeat Motif | Primer Sequence (5'–3') | Size Range (bp) | NA | HO | HE | FIS | Accession No. |
|---|---|---|---|---|---|---|---|---|
| Nsp_09 | (CTTT)3CTTC(TTCC)10TCCCTTCA(TTCC)4 | F: AGGAGGCTTTCATGTTTTCC | 128–274 | 11 | 0.533 | 0.833 | 0.360 ** | AB971583 |
| R: U19-AAATGCGTGAGAGTGAAAGG | ||||||||
| Nsp_11 †† | (GTGA)12 | F: CACTCCTTTCGCGATTCC | 298–498 | 19 | 0.692 | 0.935 | 0.259 * | AB971584 |
| R: U19-CTACCAGGTGGAGCGTGC | ||||||||
| Nsp_21 | (ACACG)11 | F: TGAAGCAAGCAATGATAAGC | 122–174 | 8 | 0.667 | 0.833 | 0.200 | AB971585 |
| R: U19-TGTTGCGTCGTGTCG | ||||||||
| Nsp_23 | (GCAC)20 | F: AACCGGGTTACCCAAAGG | 183–373 | 16 | 0.867 | 0.913 | 0.051 | AB971586 |
| R: U19-TGTGCTGACGGATGTGC | ||||||||
| Nsp_37 | (AGA)11 | F: U19-CACCCGAGACTTCGATGC | 158–167 | 3 | 0.467 | 0.504 | 0.075 | AB971587 |
| R: TGGGATGAATAAGAGCTGCC | ||||||||
| Nsp_52 | (ATC)10 | F: CTATACTGGTCGATGCGCC | 125–128 | 2 | 0.733 | 0.500 | −0.467 | AB971588 |
| R: U19-CCACTTTTGGAGTGCATGG | ||||||||
| Nsp_60 | (TCG)10 | F: GGATCCGTGTCCCTTATGC | 184–202 | 5 | 0.400 | 0.556 | 0.280 | AB971589 |
| R: U19-TAACTTCAGGGCGCTTCG | ||||||||
| Nsp_68 | (GTA)7 | F: U19-CTCGTGGGAACCCATCC | 93–112 | 6 | 1.000 | 0.758 | 0.320 | AB971590 |
| R: TCTAAACTCGCGCAAGCC | ||||||||
| Nsp_70 | (CAC)7(AAC)2 | F: U19-CCTCAGTCTGCACACCC | 102–114 | 5 | 0.400 | 0.564 | 0.291 | AB971591 |
| R: TGGAGGCGATGAAGATGG | ||||||||
| Nsp_73 | (AGC)3AAC(AGC)3(AAC)2AGCAAC(AGC)6(AAC)7(AGC)3 | F: ATGTGGGTCGGTCTCAGC | 122–137 | 6 | 0.733 | 0.704 | −0.041 | AB971592 |
| R: U19-TGCATTTGATGTTGCTGC | ||||||||
| Nsp_80 † | (CTA)7 | F: U19-TCTGGAACCGGTCTCACC | 128–134 | 3 | 0.286 | 0.349 | 0.182 | AB971593 |
| R: AATAATCCAGAGCGGGACG | ||||||||
| Nsp_81 | (ACC)7 | F: U19-CGCATAATGACAAACGC | 178–187 | 4 | 0.467 | 0.496 | 0.058 | AB971594 |
| R: CACTGAACATGCAAGCCG |
| Locus | Succeed | Size Range (bp) | NA | HO | HE | FIS |
|---|---|---|---|---|---|---|
| Nsp_09 | 18/19 | 144–177 | 7 | 0.556 | 0.682 | 0.186 |
| Nsp_21 | 16/19 | 122–137 | 3 | 0.063 | 0.174 | 0.640 ** |
| Nsp_37 | 19/19 | 161–220 | 9 | 0.737 | 0.855 | 0.138 |
| Nsp_52 | 16/19 | 119–134 | 5 | 0.438 | 0.604 | 0.275 *** |
| Nsp_60 | 18/19 | 160–196 | 4 | 0.278 | 0.335 | 0.171 |
| Nsp_68 | 14/19 | 93–118 | 8 | 0.786 | 0.747 | −0.051 |
| Nsp_70 | 18/19 | 108–117 | 4 | 0.667 | 0.500 | −0.333 |
| Nsp_73 | 18/19 | 110–125 | 6 | 0.611 | 0.645 | 0.053 |
| Nsp_81 | 19/19 | 178–187 | 3 | 0.316 | 0.476 | 0.337 |
3. Experimental Section

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Adams, D.K.; Arellano, S.M.; Govenar, B. Larval dispersal: Vent life in the water column. Oceanography 2012, 25, 256–268. [Google Scholar] [CrossRef]
- Newman, W.A.; Hessler, R.R. A new abyssal hydrothermal verrucomorpha (Cirripedia; Sessilia): The most primitive living sessile barnacle. Trans. San Diego Soc. Nat. Hist. 1989, 21, 256–273. [Google Scholar]
- Newman, W.A. A new genus and species of barnacle (Cirripedia, Verrucomorpha) associated with vents of the Lau Back-Arc Basin: Its gross morphology, inferred first juvenile stage and affinities. Zoosystema 2000, 22, 71–84. [Google Scholar]
- Watanabe, H. Dispersal and evolution in chemoautosynthesis-based communities in the western Pacific: Verrucomorphs as test species for evolutionary studies on hydrothermal vent-endemic animals. Jpn. J. Benthol. 2003, 58, 44–49. [Google Scholar]
- Watanabe, H.; Tsuchida, S.; Fujikura, K.; Yamamoto, H.; Inagaki, F.; Kyo, M.; Kojima, S. Population history associated with hydrothermal vent activity inferred from genetic structure of neoverrucid barnacles around Japan. Mar. Ecol. Prog. Ser. 2005, 288, 233–240. [Google Scholar] [CrossRef]
- Newman, W.A. Juvenile ontogeny and metamorphosis in the most primitive living sessile barnacle, Neoverruca, from abyssal hydrothermal springs. Bull. Mar. Sci. 1989, 45, 467–477. [Google Scholar]
- Aronesty, E. Ea-Utils: “Command-Line Tools for Processing Biological Sequencing Data”. 2011. Available online: http://code.google.com/p/ea-utils (accessed on 19 November 2013).
- Castoe, T.A.; Poole, A.W.; de Koning, A.P.; Jones, K.L.; Tomback, D.F.; Oyler-Mccance, S.J.; Fike, J.A.; Lance, S.L.; Streicher, J.W.; Smith, E.N.; et al. Rapid microsatellite identification from Illumina paired-end genomic sequencing in two birds and a snake. PLoS One 2012, 7, e30953. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar]
- Van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. Micro-Checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. Genepop (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar]
- Rousset, F. Genepop’007: A complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Genepop on the web. Available online: http://genepop.curtin.edu.au/ (accessed on 5 April 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakajima, Y.; Shinzato, C.; Khalturina, M.; Watanabe, H.; Inagaki, F.; Satoh, N.; Mitarai, S. Cross-Species, Amplifiable Microsatellite Markers for Neoverrucid Barnacles from Deep-Sea Hydrothermal Vents Developed Using Next-Generation Sequencing. Int. J. Mol. Sci. 2014, 15, 14364-14371. https://doi.org/10.3390/ijms150814364
Nakajima Y, Shinzato C, Khalturina M, Watanabe H, Inagaki F, Satoh N, Mitarai S. Cross-Species, Amplifiable Microsatellite Markers for Neoverrucid Barnacles from Deep-Sea Hydrothermal Vents Developed Using Next-Generation Sequencing. International Journal of Molecular Sciences. 2014; 15(8):14364-14371. https://doi.org/10.3390/ijms150814364
Chicago/Turabian StyleNakajima, Yuichi, Chuya Shinzato, Mariia Khalturina, Hiromi Watanabe, Fumio Inagaki, Nori Satoh, and Satoshi Mitarai. 2014. "Cross-Species, Amplifiable Microsatellite Markers for Neoverrucid Barnacles from Deep-Sea Hydrothermal Vents Developed Using Next-Generation Sequencing" International Journal of Molecular Sciences 15, no. 8: 14364-14371. https://doi.org/10.3390/ijms150814364