Neuroprotective Effects of Sulforaphane on Cholinergic Neurons in Mice with Alzheimer’s Disease-Like Lesions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
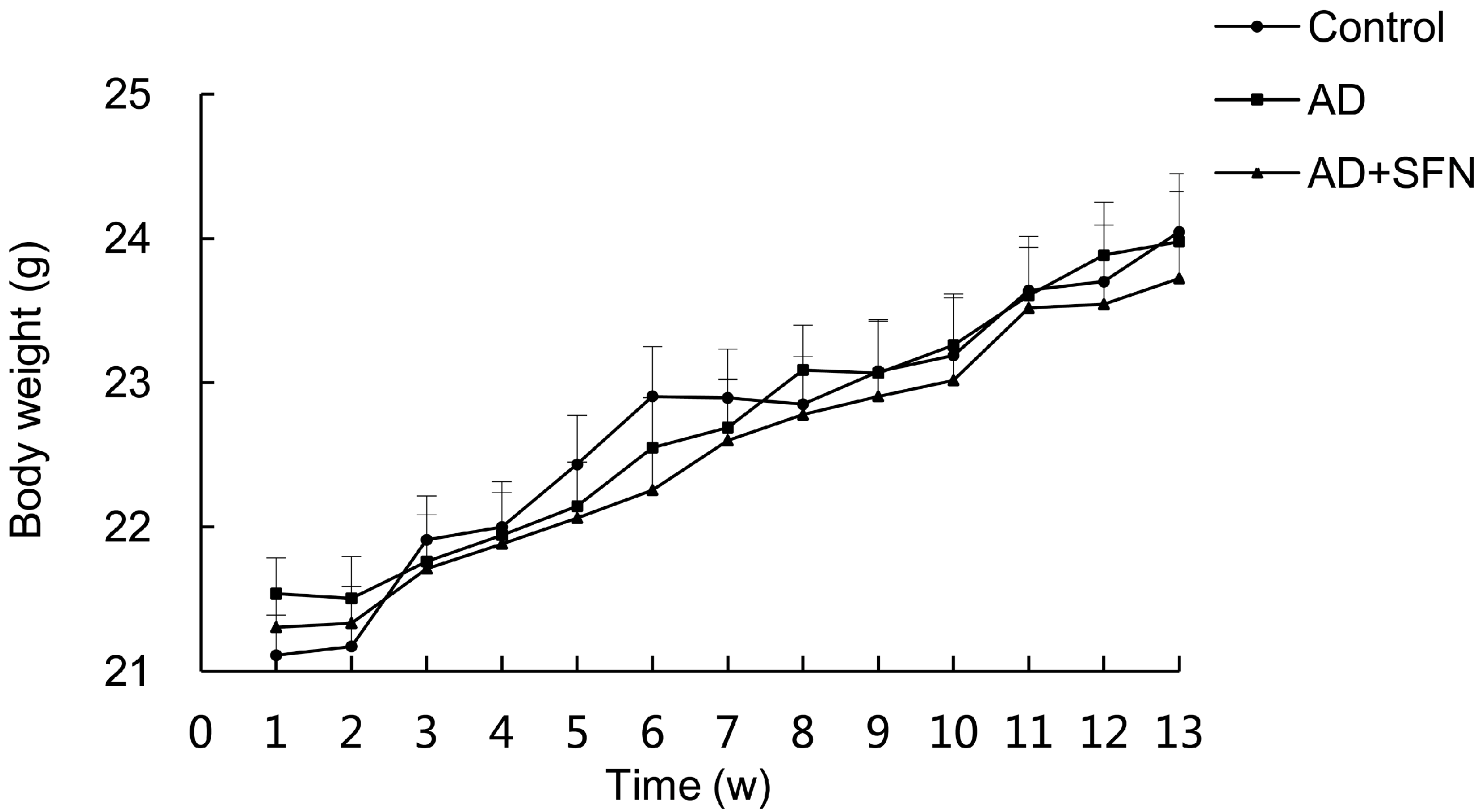
2.1. Signs and Body Weight

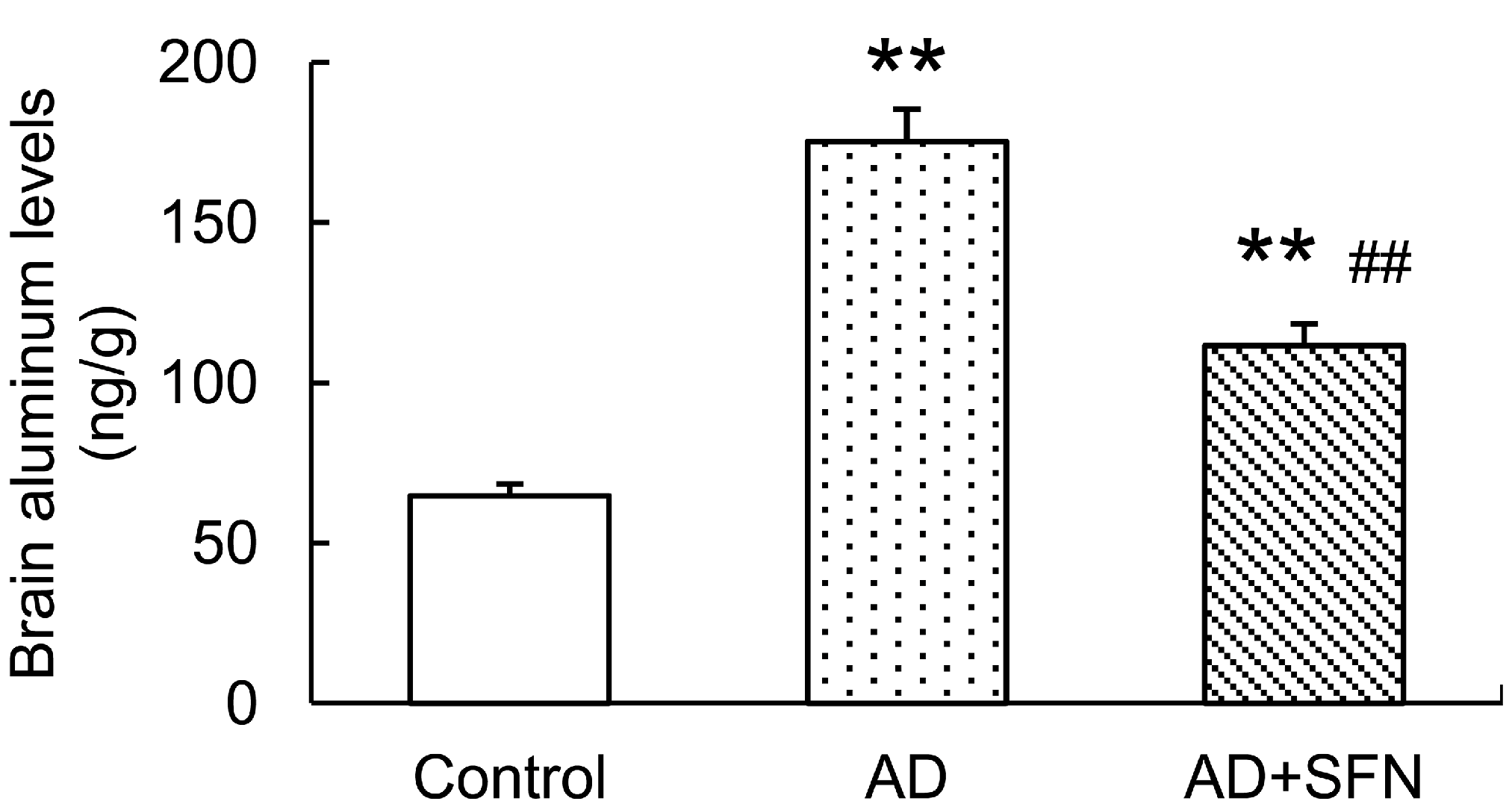
2.2. Analysis of Aluminum Level in the Mouse Brain

2.3. Step-Down-Type Passive Avoidance Tests

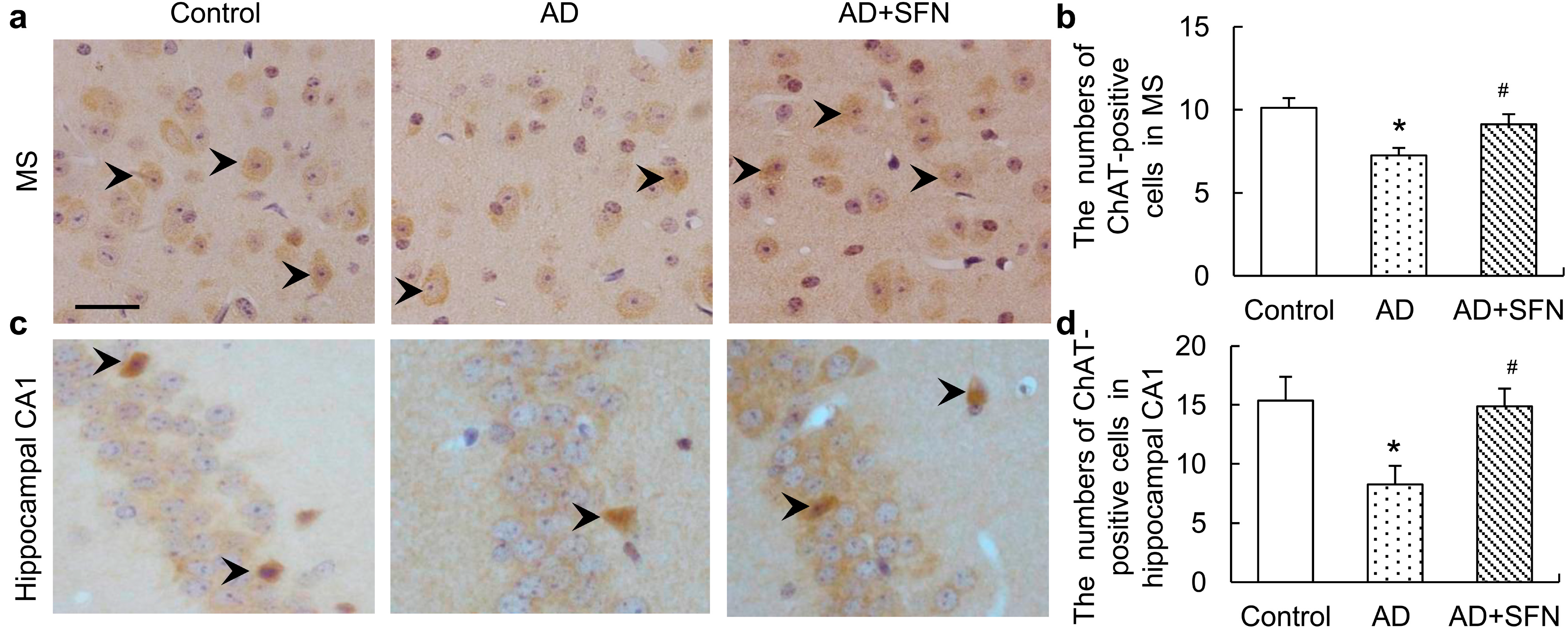
2.4. Choline Acetyltransferase (ChAT) Immuno-Positive Neuron Assays

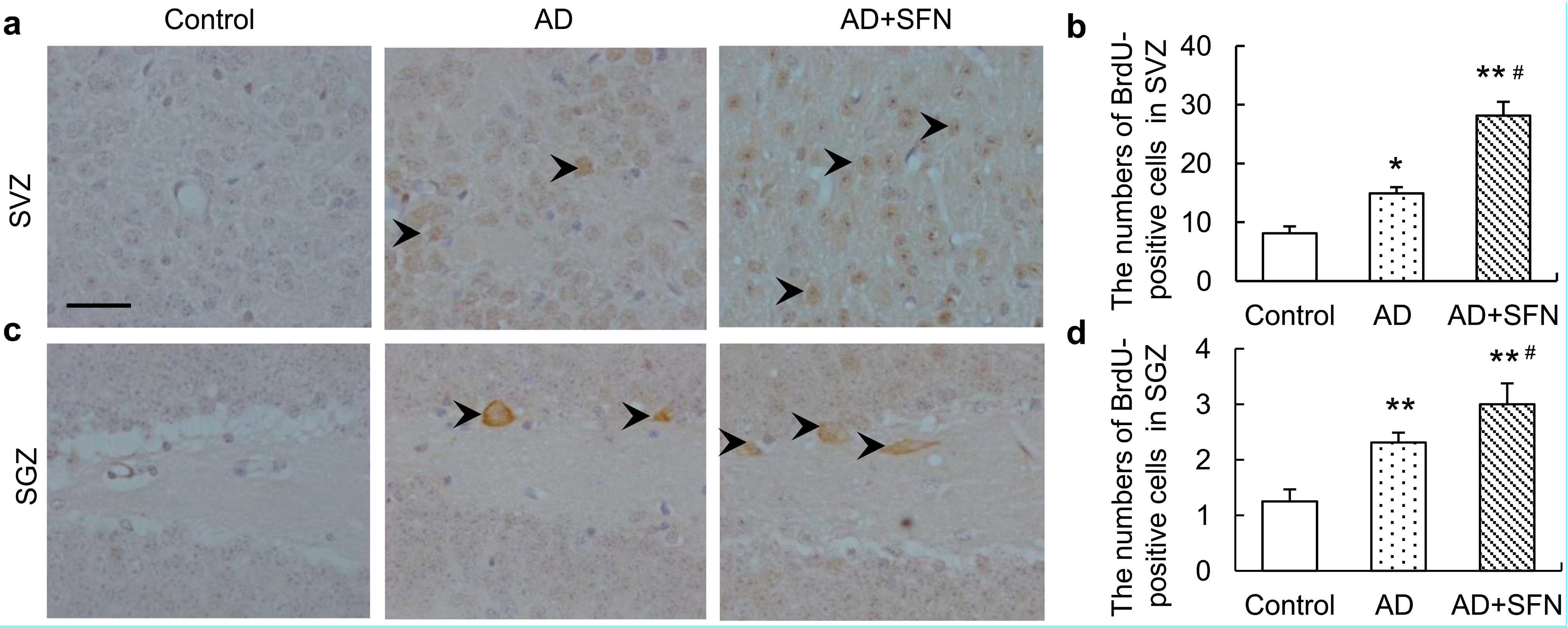
2.5. 5-Bromo-2'-deoxyuridine Immuno-Positive Cell Assays
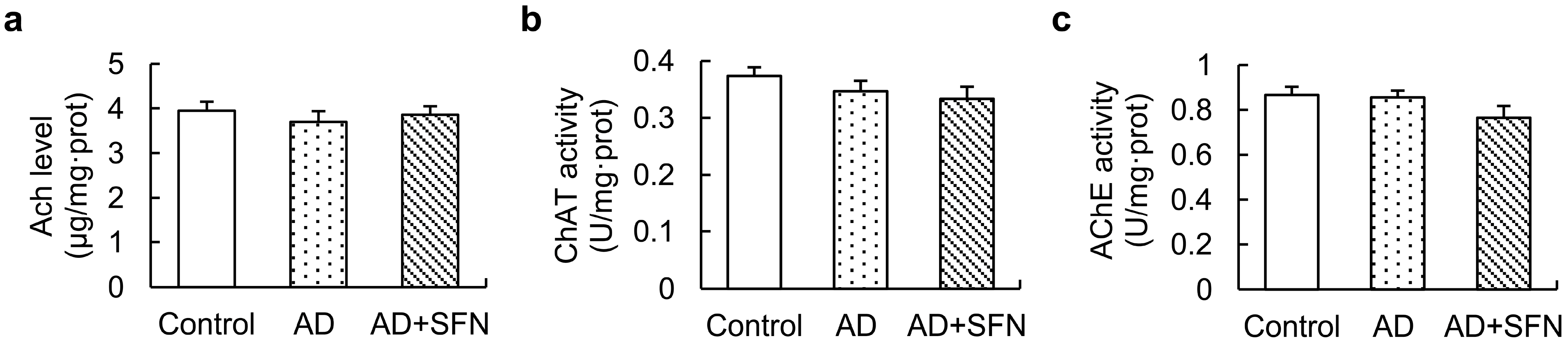
2.6. Acetylcholine (Ach) Level and Activities of ChAT and Acetylcholinesterase in the Cerebral Cortex


2.7. Discussion
3. Experimental Section
3.1. Reagents
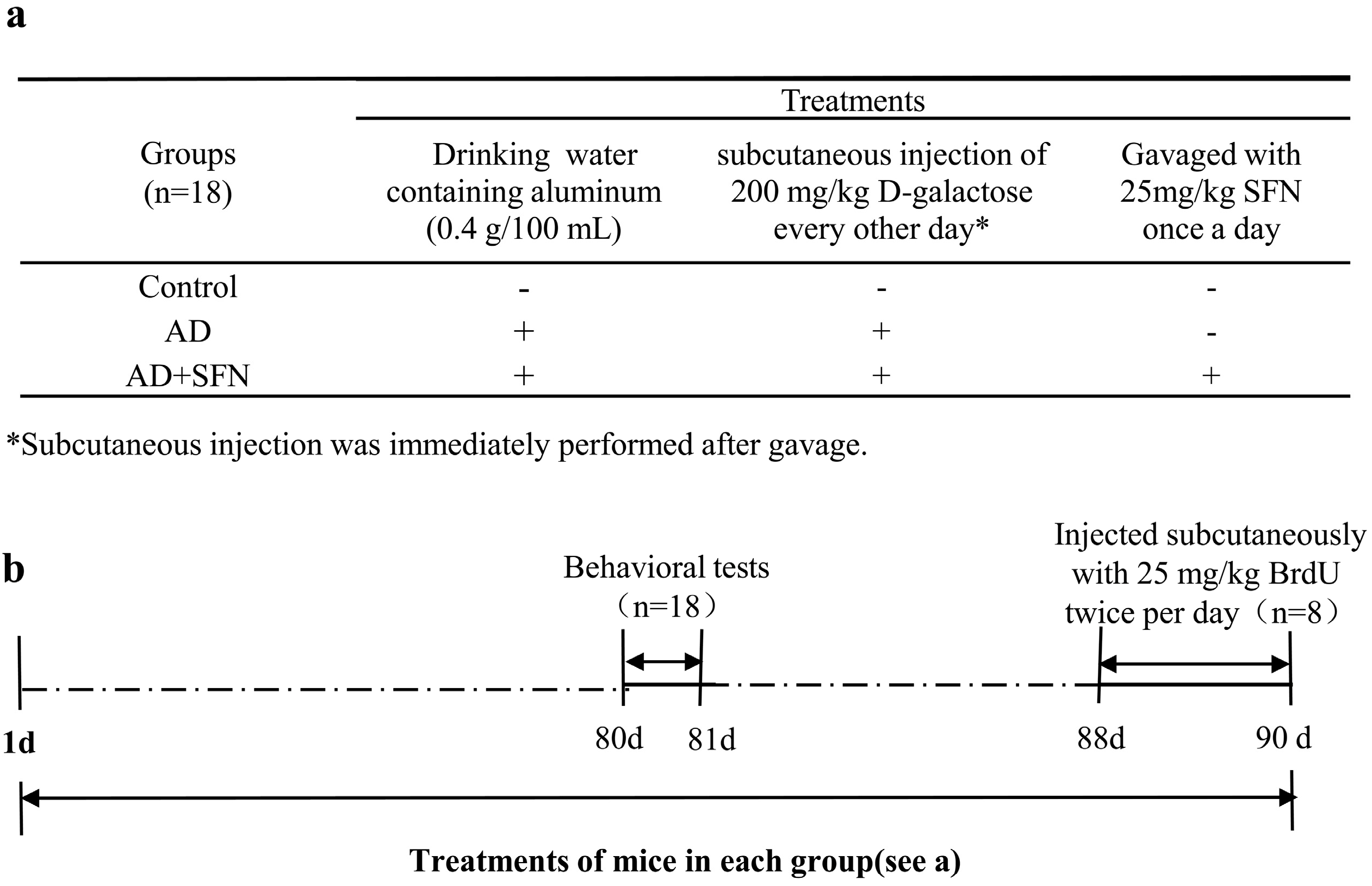
3.2. Animals, Treatments, and Tissue Collection

3.3. Step-Down-Type Passive Avoidance Tests
3.4. Aluminum Measurement by Atomic Absorption Spectrometry
3.5. Immunohistochemistry Assays
3.6. ACh Level and Activities of ChAT and AChE
3.7. Statistical Analyses
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Wimo, A.; Winblad, B.; Aguero-Torres, H.; von Strauss, E. The magnitude of dementia occurrence in the world. Alzheimer Dis. Assoc. Disord. 2003, 17, 63–67. [Google Scholar] [CrossRef]
- Alagiakrishnan, K.; Gill, S.S.; Fagarasanu, A. Genetics and epigenetics of Alzheimer’s disease. Postgrad. Med. J. 2012, 88, 522–529. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Gil-Bea, F.J.; García-Alloza, M.; Domínguez, J.; Marcos, B.; Ramírez, M.J. Evaluation of cholinergic markers in Alzheimer’s disease and in a model of cholinergic deficit. Neurosci. Lett. 2005, 375, 37–41. [Google Scholar] [CrossRef]
- Van Marum, R.J. Current and future therapy in Alzheimer’s disease. Fundam. Clin. Pharmacol. 2008, 22, 265–274. [Google Scholar] [CrossRef]
- Lleó, A; Greenberg, S.M.; Growdon, J.H. Current pharmacotherapy for Alzheimer’s disease. Annu. Rev. Med. 2006, 57, 513–533. [Google Scholar] [CrossRef]
- Howes, M.J.; Perry, E. The role of phytochemicals in the treatment and prevention of dementia. Drugs Aging 2011, 28, 439–468. [Google Scholar] [CrossRef]
- Lee, C.; Park, G.H.; Lee, S.R.; Jang, J.H. Attenuation of β-amyloid-induced oxidative cell death by sulforaphane via activation of NF-E2-related factor 2. Oxid. Med. Cell. Longev. 2013. [Google Scholar] [CrossRef]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef]
- Xiao, F.; Li, X.G.; Zhang, X.Y.; Hou, J.D.; Lin, L.F.; Gao, Q.; Luo, H.M. Combined administration of d-galactose and aluminium induces Alzheimer-like lesions in brain. Neurosci. Bull. 2011, 27, 143–155. [Google Scholar] [CrossRef]
- Zhang, R.; Miao, Q.W.; Zhu, C.X.; Zhao, Y.; Liu, L.; Yang, J.; An, L. Sulforaphane ameliorates neurobehavioral deficits and protects the brain from amyloid β deposits and peroxidation in mice with Alzheimer-like lesions. Am. J. Alzheimer’s Dis. Other Demen. 2014. [Google Scholar] [CrossRef]
- Musilli, M.; Nicolia, V.; Borrelli, S.; Scarpa, S.; Diana, G. Behavioral effects of Rho GTPase modulation in a model of Alzheimer’s disease. Behav. Brain Res. 2013, 237, 223–229. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar]
- Fraser, D.D.; MacVicar, B.A. Cholinergic-dependent plateau potential in hippocampal CA1 pyramidal neurons. J. Neurosci. 1996, 16, 4113–4128. [Google Scholar]
- Gu, H.; Long, D.; Song, C.; Li, X. Recombinant human NGF-loaded microspheres promote survival of basal forebrain cholinergic neurons and improve memory impairments of spatial learning in the rat model of Alzheimer’s disease with fimbria-fornix lesion. Neurosci. Lett. 2009, 453, 204–209. [Google Scholar] [CrossRef]
- Li, Q.; Chen, M.; Liu, H.; Yang, L.; Yang, G. Expression of APP, BACE1, AChE and ChAT in an AD model in rats and the effect of donepezil hydrochloride treatment. Mol. Med. Rep. 2012, 6, 1450–1455. [Google Scholar]
- Scali, C.; Giovannini, M.G.; Prosperi, C.; Bellucci, A.; Pepeu, G.; Casamenti, F. The selective cyclooxygenase-2 inhibitor rofecoxib suppresses brain inflammation and protects cholinergic neurons from excitotoxic degeneration in vivo. Neuroscience 2003, 117, 909–919. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, S.; Wang, L. The impact of Boschiakia Rossica extract on the Ach, ChAT and AChE of AD rats. Chin. Arch. Trad. Chin. Med. 2009, 27, 1874–1876. (In Chinese) [Google Scholar]
- Giacobini, E. Cholinergic function and Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2003, 18, S1–S5. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Ikonomovic, M.D.; Styren, S.D.; Beckett, L.; Wisniewski, S.; Bennett, D.A.; Cochran, E.J.; Kordower, J.H.; Mufson, E.J. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol. 2002, 51, 145–155. [Google Scholar] [CrossRef]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane: Translational research from laboratory bench to clinic. Nutr. Rev. 2013, 71, 709–726. [Google Scholar] [CrossRef]
- Alfieri, A.; Srivastava, S.; Siow, R.C.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood-brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef]
- Dash, P.K.; Zhao, J.; Orsi, S.A.; Zhang, M.; Moore, A.N. Sulforaphane improves cognitive function administered following traumatic brain injury. Neurosci. Lett. 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Morroni, F.; Tarozzi, A.; Sita, G.; Bolondi, C.; Zolezzi-Moraga, J.M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson’s disease. Neurotoxicology 2013, 36, 63–71. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, J.; Yu, S.; Chen, Y.; Wu, J.; Zhao, Y. Sulforaphane protects primary cultures of cortical neurons against injury induced by oxygen-glucose deprivation/reoxygenation via antiapoptosis. Neurosci. Bull. 2012, 28, 509–516. [Google Scholar] [CrossRef]
- Wang, X.; de Rivero Vaccari, J.P.; Wang, H.; Diaz, P.; German, R.; Marcillo, A.E.; Keane, R.W. Activation of the nuclear factor E2-related factor 2/antioxidant response element pathway is neuroprotective after spinal cord injury. J. Neurotrauma 2012, 29, 936–945. [Google Scholar] [CrossRef]
- Goldman, S. Glia as neural progenitor cells. Trends Neurosci. 2003, 26, 590–596. [Google Scholar] [CrossRef]
- Alvarez-Buylla, A.; García-Verdugo, J.M.; Tramontin, A.D. A unified hypothesis on the lineage of neural stem cells. Nat. Rev. Neurosci. 2001, 2, 287–293. [Google Scholar] [CrossRef]
- Rockenstein, E.; Ubhi, K.; Doppler, E.; Novak, P.; Moessler, H.; Li, B.; Blanchard, J.; Grundke-Iqbal, I.; Iqbal, K.; Mante, M.; et al. Regional comparison of the neurogenic effects of CNTF-derived peptides and cerebrolysin in AβPP transgenic mice. J. Alzheimer’s Dis. 2011, 27, 743–752. [Google Scholar]
- Lilja, A.M.; Röjdner, J.; Mustafiz, T.; Thomé, C.M.; Storelli, E.; Gonzalez, D.; Unger-Lithner, C.; Greig, N.H.; Nordberg, A.; Marutle, A. Age-dependent neuroplasticity mechanisms in Alzheimer Tg2576 mice following modulation of brain amyloid-β levels. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Choi, H.; Park, H.H.; Lee, K.Y.; Choi, N.Y.; Yu, H.J.; Lee, Y.J.; Park, J.; Huh, Y.M.; Lee, S.H.; Koh, S.H. Coenzyme Q10 restores amyloid beta-inhibited proliferation of neural stem cells by activating the PI3K pathway. Stem Cells Dev. 2013, 22, 2112–2120. [Google Scholar] [CrossRef]
- Hu, Y.S.; Xu, P.; Pigino, G.; Brady, S.T.; Larson, J.; Lazarov, O. Complex environment experience rescues impaired neurogenesis, enhances synaptic plasticity, and attenuates neuropathology in familial Alzheimer’s disease-linked APPswe/PS1DeltaE9 mice. FASEB J. 2010, 24, 1667–1681. [Google Scholar] [CrossRef]
- Kamphuis, W.; Orre, M.; Kooijman, L.; Dahmen, M.; Hol, E.M. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia 2012, 60, 615–629. [Google Scholar] [CrossRef]
- Díaz-Moreno, M.; Hortigüela, R.; Gonçalves, A.; García-Carpio, I.; Manich, G.; García-Bermúdez, E.; Moreno-Estellés, M.; Eguiluz, C.; Vilaplana, J.; Pelegrí, C.; et al. Aβ increases neural stem cell activity in senescence-accelerated SAMP8 mice. Neurobiol. Aging 2013, 34, 2623–2638. [Google Scholar] [CrossRef]
- Kanematsu, S.; Yoshizawa, K.; Uehara, N.; Miki, H.; Sasaki, T.; Kuro, M.; Lai, Y.C.; Kimura, A.; Yuri, T.; Tsubura, A. Sulforaphane inhibits the growth of KPL-1 human breast cancer cells in vitro and suppresses the growth and metastasis of orthotopically transplanted KPL-1 cells in female athymic mice. Oncol. Rep. 2011, 26, 603–608. [Google Scholar]
- Li, S.H.; Fu, J.; Watkins, D.N.; Srivastava, R.K.; Shankar, S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog-GLI pathway. Mol. Cell. Biochem. 2013, 373, 217–227. [Google Scholar] [CrossRef]
- Chen, H.; Landen, C.N.; Li, Y.; Alvarez, R.D.; Tollefsbol, T.O. Epigallocatechin gallate and sulforaphane combination treatment induce apoptosis in paclitaxel-resistant ovarian cancer cells through hTERT and Bcl-2 down-regulation. Exp. Cell Res. 2013, 319, 697–706. [Google Scholar] [CrossRef]
- Zanichelli, F.; Capasso, S.; Cipollaro, M.; Pagnotta, E.; Cartenì, M.; Casale, F.; Iori, R.; Galderisi, U. Dose-dependent effects of R-sulforaphane isothiocyanate on the biology of human mesenchymal stem cells, at dietary amounts, it promotes cell proliferation and reduces senescence and apoptosis, while at anti-cancer drug doses, it has a cytotoxic effect. Age 2012, 34, 281–293. [Google Scholar] [CrossRef]
- La Marca, M.; Beffy, P.; Della Croce, C.; Gervasi, P.G.; Iori, R.; Puccinelli, E.; Longo, V. Structural influence of isothiocyanates on expression of cytochrome P450, phase II enzymes, and activation ofNrf2 in primary rat hepatocytes. Food Chem. Toxicol. 2012, 50, 2822–2830. [Google Scholar] [CrossRef] [Green Version]
- Angeloni, C.; Leoncini, E.; Malaguti, M.; Angelini, S.; Hrelia, P.; Hrelia, S. Modulation of phase II enzymes by sulforaphane: Implications for its cardioprotective potential. J. Agric. Food Chem. 2009, 57, 5615–5622. [Google Scholar] [CrossRef]
- James, B.D.; Bennett, D.A.; Boyle, P.A.; Leurgans, S.; Schneider, J.A. Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA 2012, 307, 1798–1800. [Google Scholar] [CrossRef]
- Vom Berg, J.; Prokop, S.; Miller, K.R.; Obst, J.; Kälin, R.E.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819. [Google Scholar] [CrossRef]
- Paul, M.K.; Bisht, B.; Darmawan, D.O.; Chiou, R.; Ha, V.L.; Wallace, W.D.; Chon, A.T.; Hegab, A.E.; Grogan, T.; Elashoff, D.A.; et al. Dynamic changes in intracellular ROS levels regulate airway basal stem cell homeostasis through Nrf2-dependent notch signaling. Cell Stem Cell 2014. [Google Scholar] [CrossRef]
- Hoetzenecker, W.; Echtenacher, B.; Guenova, E.; Hoetzenecker, K.; Woelbing, F.; Brück, J.; Teske, A.; Valtcheva, N.; Fuchs, K.; Kneilling, M.; et al. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat. Med. 2011, 18, 128–134. [Google Scholar] [CrossRef]
- Ukai, M.; Watanabe, Y.; Kameyama, T. Endomorphins 1 and 2, endogenous mu-opioid receptor agonists, impair passive avoidance learning in mice. Eur. J. Pharmacol. 2001, 421, 115–119. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Koseki, M.; Wakamatsu, M.; Matsumura, E. Effects of systemic administration of beta-casomorphin-5 on learning and memory in mice. Eur. J. Pharmacol. 2006, 530, 81–87. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, R.; Zhang, J.; Fang, L.; Li, X.; Zhao, Y.; Shi, W.; An, L. Neuroprotective Effects of Sulforaphane on Cholinergic Neurons in Mice with Alzheimer’s Disease-Like Lesions. Int. J. Mol. Sci. 2014, 15, 14396-14410. https://doi.org/10.3390/ijms150814396
Zhang R, Zhang J, Fang L, Li X, Zhao Y, Shi W, An L. Neuroprotective Effects of Sulforaphane on Cholinergic Neurons in Mice with Alzheimer’s Disease-Like Lesions. International Journal of Molecular Sciences. 2014; 15(8):14396-14410. https://doi.org/10.3390/ijms150814396
Chicago/Turabian StyleZhang, Rui, Jingzhu Zhang, Lingduo Fang, Xi Li, Yue Zhao, Wanying Shi, and Li An. 2014. "Neuroprotective Effects of Sulforaphane on Cholinergic Neurons in Mice with Alzheimer’s Disease-Like Lesions" International Journal of Molecular Sciences 15, no. 8: 14396-14410. https://doi.org/10.3390/ijms150814396