Simvastatin Attenuates the Oxidative Stress, Endothelial Thrombogenicity and the Inducibility of Atrial Fibrillation in a Rat Model of Ischemic Heart Failure

Abstract
:1. Introduction
2. Results and Discussion
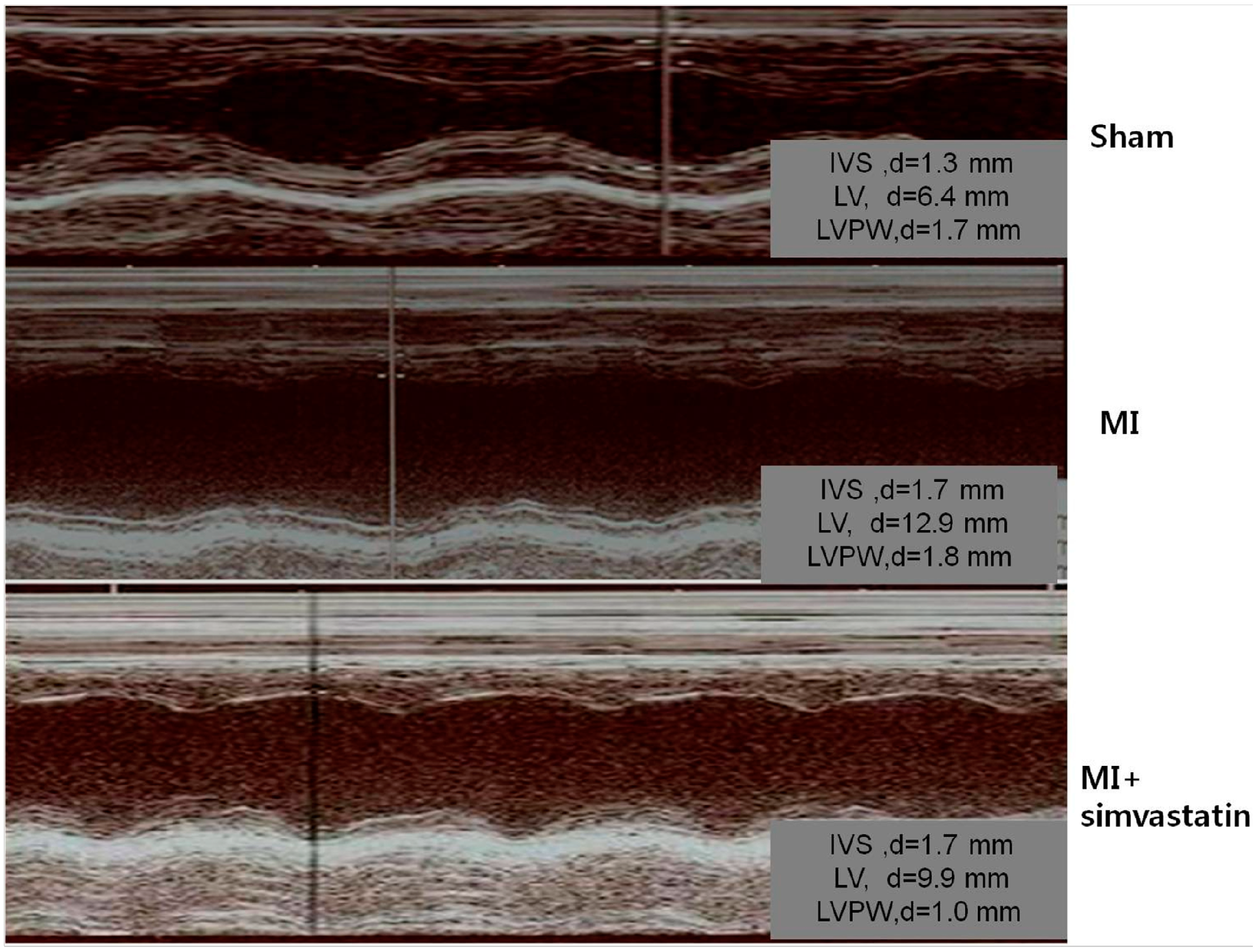
2.1. Echocardiographic Indices


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Sham ( n = 10) | MI ( n = 10) | MI-Simvastatin ( n = 10) |
|---|---|---|---|
| EF, % | 83.0 ± 7.6 | 30.0 ± 16.1 * | 43.0 ± 21.5 * |
| FS, % | 48.5 ± 8.8 | 12.7 ± 7.5 * | 20.6 ± 15.2 * |
| LVEDD, mm | 7.9 ± 0.6 | 9.5 ± 1.3 ** | 9.6 ± 1.6 *** |
| LVESD, mm | 4.0 ± 0.8 | 8.3 ± 1.5 * | 7.8 ± 2.2 * |
| LAD, mm | 3.4 ± 0.8 | 3.8 ± 0.9 | 3.6 ± 0.8 |
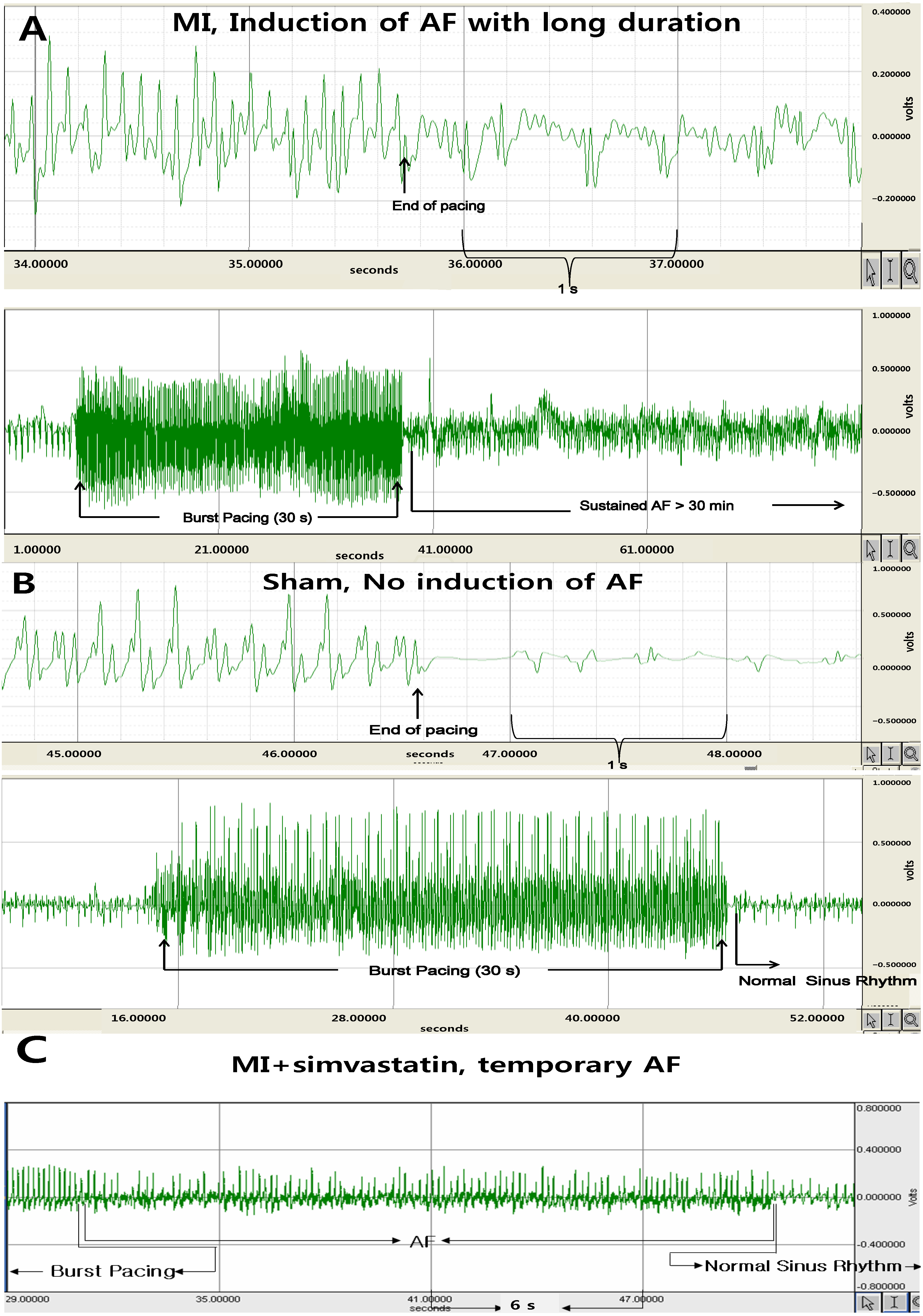
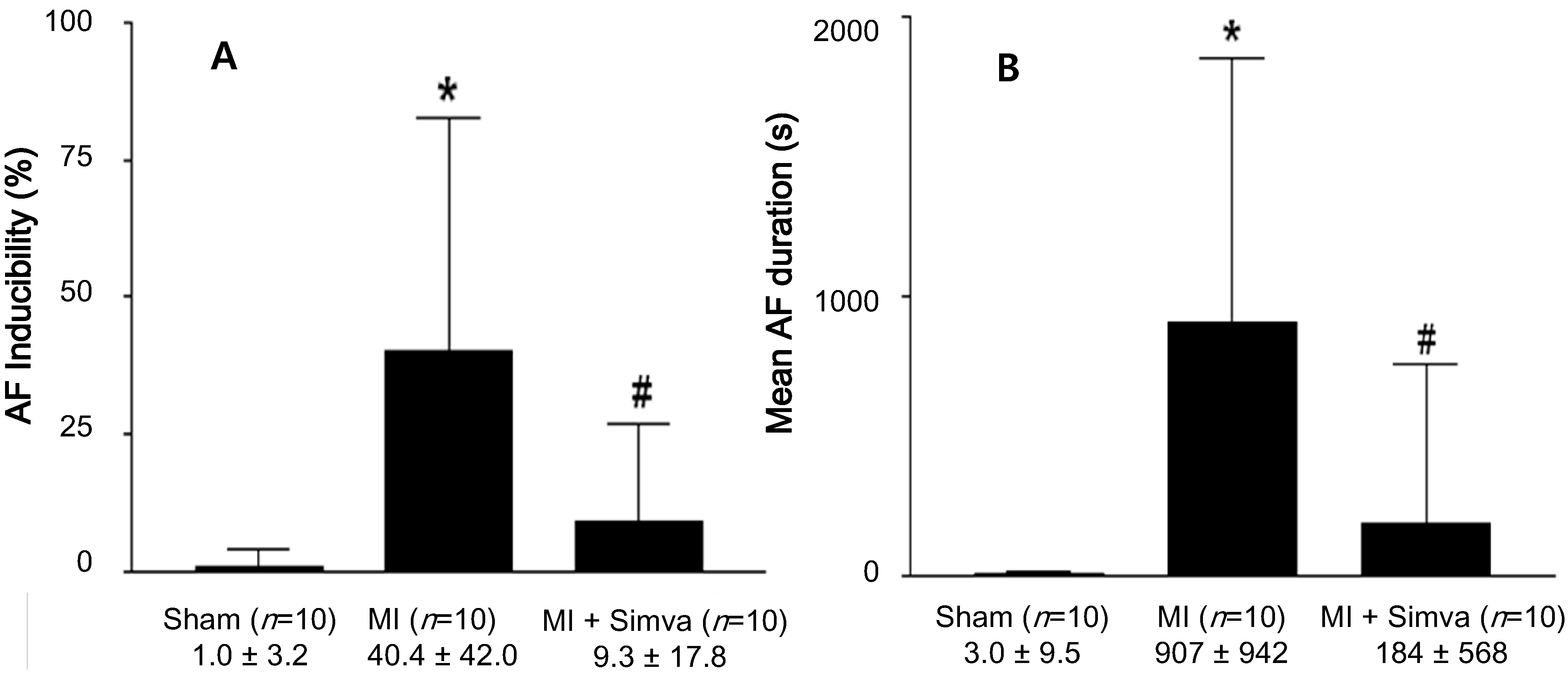
2.2. Atrial Fibrillation Induction Study

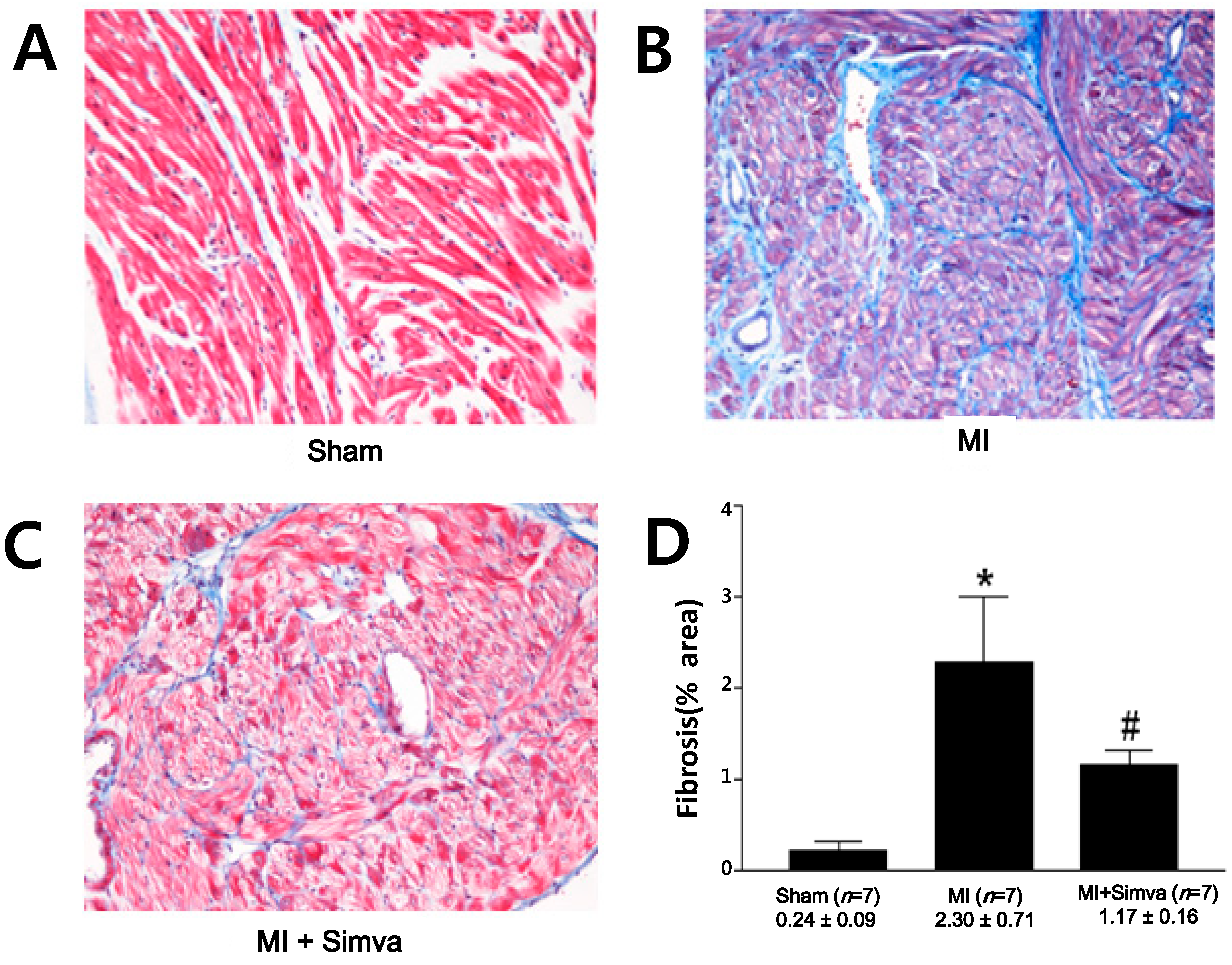
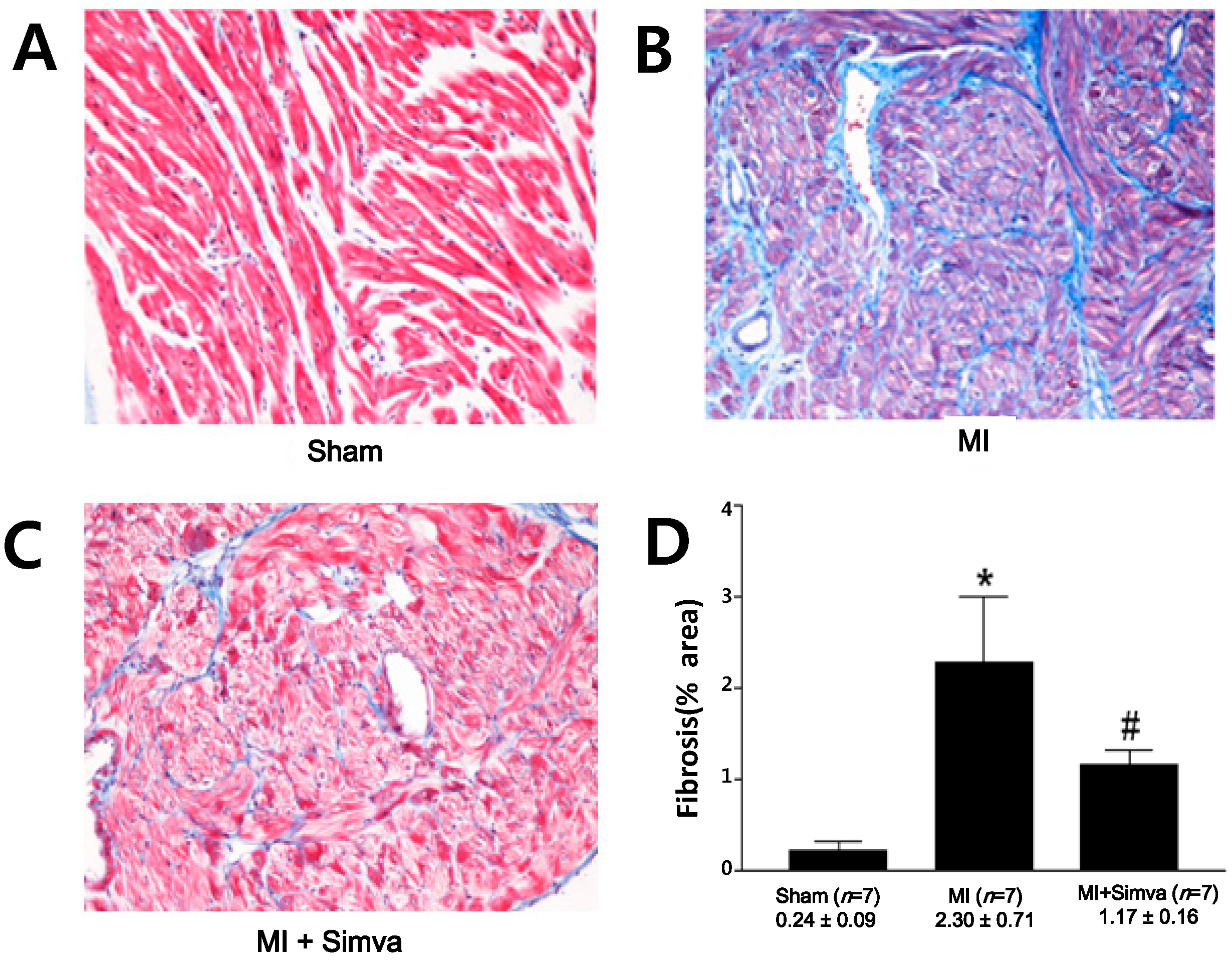
2.3. Reduction of Atrial Fibrosis by Simvastatin

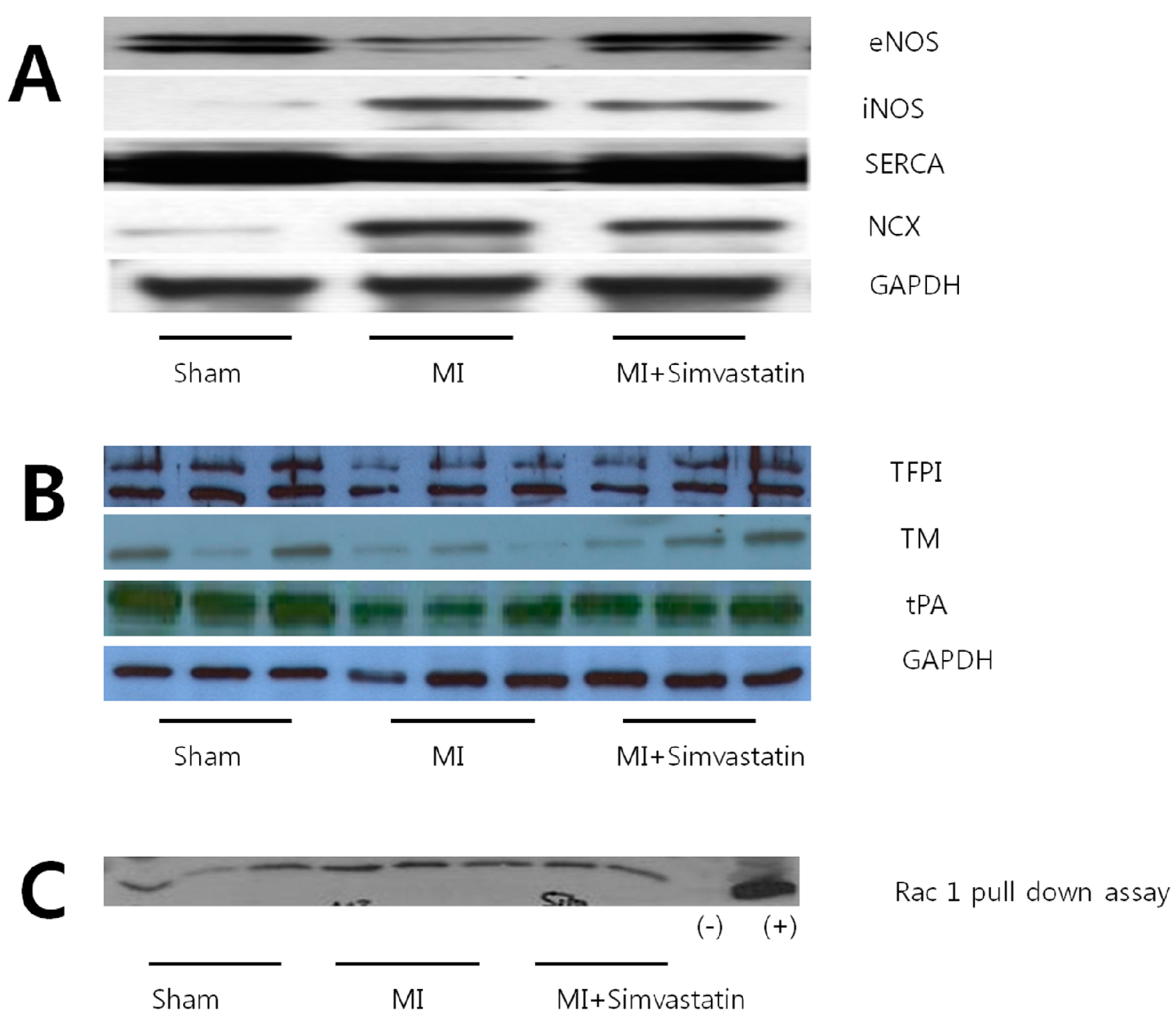
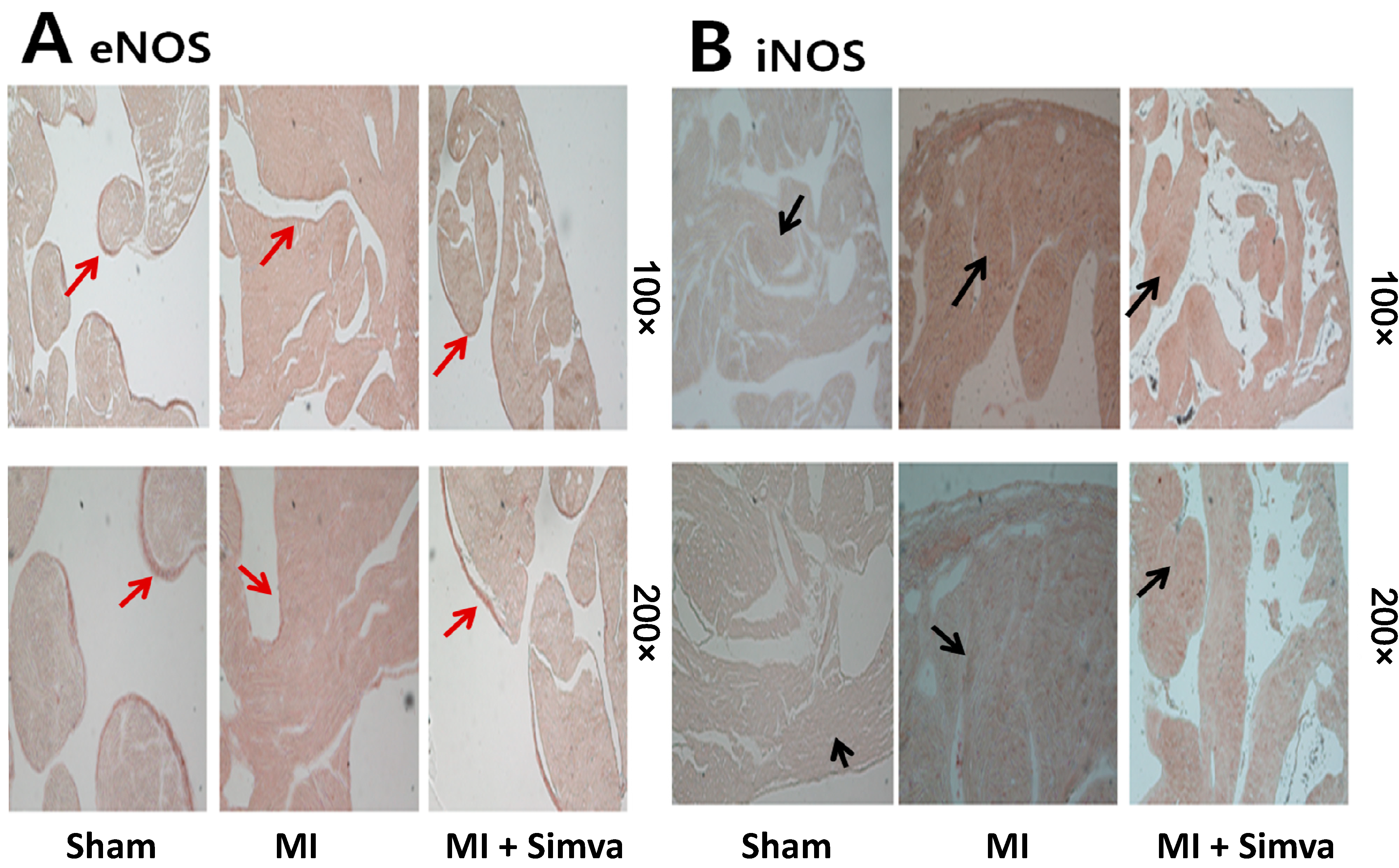
2.4. Expression of Nitric Oxide Synthases and Calcium Handling Proteins
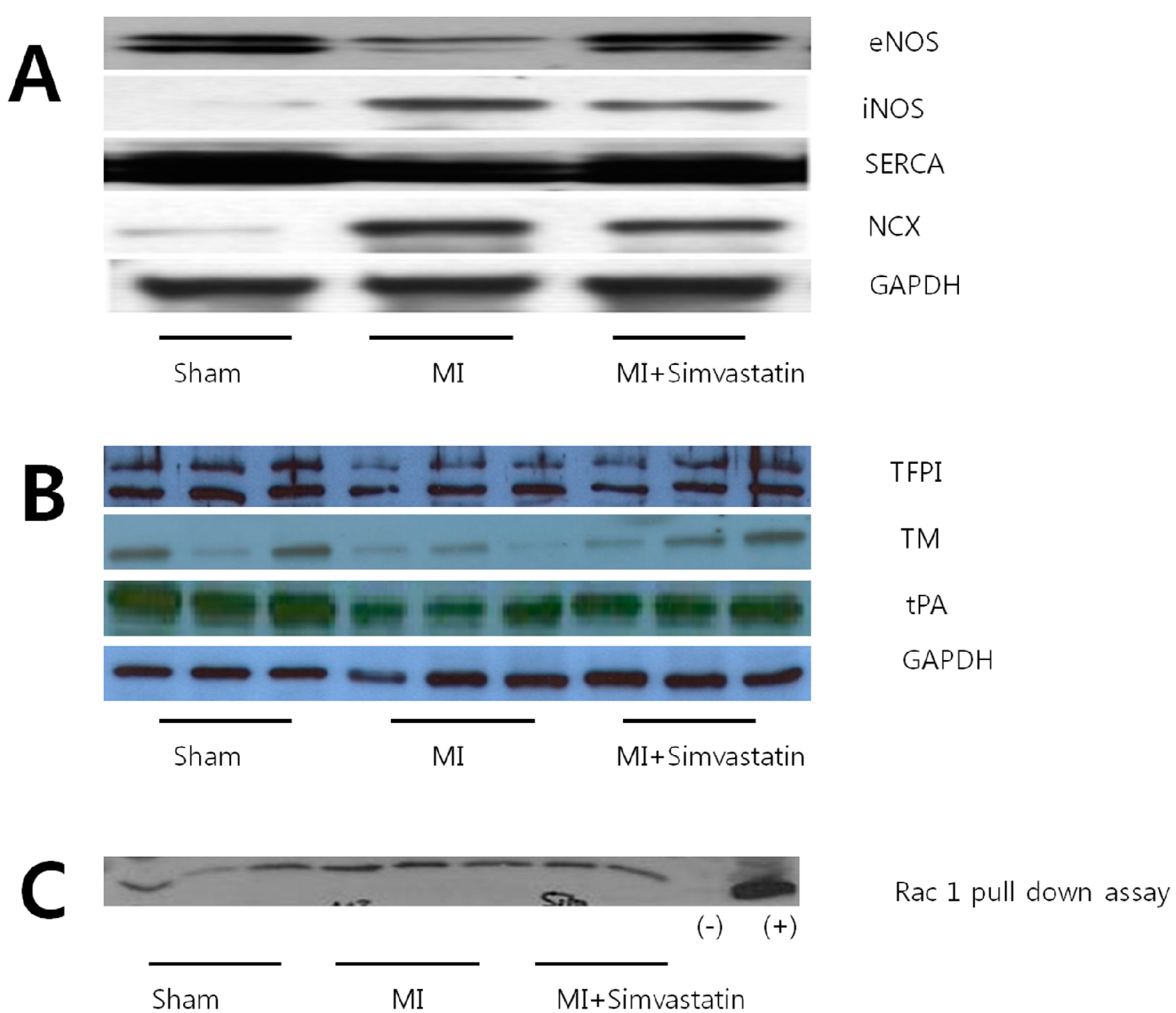
| Parameters | Sham ( n = 10) | MI ( n = 10) | MI-Simvastatin ( n = 10) |
|---|---|---|---|
| Fibrosis (%) | 0.24 ± 0.09 ( n = 7) | 2.30 ± 0.71 * ( n = 7) | 1.17 ± 0.16 # ( n = 7) |
| eNOS | 13.41 ± 2.58 ( n = 6) | 6.75 ± 2.05 * ( n = 6) | 12.06 ± 3.43 # ( n = 6) |
| iNOS | 0.28 ± 0.30 ( n = 9) | 7.33 ± 5.79 * ( n = 10) | 1.60 ± 1.47 **,# ( n = 12) |
| INCX | 0.79 ± 0.48 ( n = 8) | 14.52 ± 11.87 * ( n = 11) | 4.19 ± 11.87 *,## ( n = 12) |
| SERCA | 30.90 ± 3.18 ( n = 6) | 21.13 ± 1.36 * ( n = 6) | 25.43 ± 3.27 **,## ( n = 6) |
| TM | 2.54 ± 0.47 ( n = 6) | 0.80 ± 0.28 * ( n = 6) | 1.85 ± 0.38 # ( n = 6) |
| TFPI | 6.37 ± 0.71 ( n = 6) | 3.15 ± 0.22 ( n = 6) | 4.32 ± 0.47 ( n = 6) |
| tPA | 2.73 ± 0.12 ( n = 6) | 1.25 ± 0.12 ( n = 6) | 2.01 ± 0.13 ( n = 6) |
| Rac 1 | 16,165 ± 1696 ( n = 6) | 28,440 ± 1655 * ( n = 6) | 19,275 ± 2236 # ( n = 5) |
2.5. Immunohistochemistry Results

2.6. Thrombogenicity and Rac 1 Activity
3. Experimental Section
3.1. Animals Models
3.2. Echocardiogram
3.3. Electrophysiological Study
3.4. Quantification of Fibrosis
3.5. Western Blot
3.6. Rac1 GST-p21-Activated Kinase Pull-Down Assay
3.7. Immunohistochemistry for eNOS and iNOS
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Go, A.S.; Hylek, E.M.; Phillips, K.A.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001, 285, 2370–2375. [Google Scholar]
- Ock, S.Y.; Cho, K.I.; Kim, H.J.; Lee, N.Y.; Kim, E.J.; Kim, N.K.; Lee, W.H.; Yeo, G.E.; Heo, J.J.; Han, Y.J.; et al. The impacts of C-reactive protein and atrial fibrillation on carotid atherosclerosis and ischemic stroke in patients with suspected ischemic cerebrovascular disease: A single-center retrospective observational cohort study. Korean Circ. J. 2013, 43, 796–803. [Google Scholar] [CrossRef]
- Korantzopoulos, P.; Kolettis, T.; Siogas, K.; Goudevenos, J. Atrial fibrillation and electrical remodeling: The potential role of inflammation and oxidative stress. Med. Sci. Monit. 2003, 9, RA225–RA229. [Google Scholar]
- Cai, H.; Li, Z.; Goette, A.; Mera, F.; Honeycutt, C.; Feterik, K.; Wilcox, J.N.; Dudley, S.C.; Harrison, D.G.; Langberg, J.J. Down-regulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: Potential mechanisms for atrial thrombosis and stroke. Circulation 2002, 106, 2854–2858. [Google Scholar] [CrossRef]
- Aviles, R.J.; Martin, D.O.; Apperson-Hansen, C.; Houghtaling, P.L.; Rautaharju, P.; Kronmal, R.A.; Tracy, R.P.; van Wagoner, D.R.; Psaty, B.M.; Lauer, M.S.; et al. Inflammation as a risk factor for atrial fibrillation. Circulation 2003, 108, 3006–3010. [Google Scholar] [CrossRef]
- Maron, D.J.; Fazio, S.; Linton, M.F. Current perspectives on statins. Circulation 2000, 101, 207–213. [Google Scholar] [CrossRef]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N. Engl. J. Med. 1986, 315, 1046–1051. [Google Scholar] [CrossRef]
- Laufs, U.; Liao, J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by rho gtpase. J. Biol. Chem. 1998, 273, 24266–24271. [Google Scholar] [CrossRef]
- Maguy, A.; Hebert, T.E.; Nattel, S. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc. Res. 2006, 69, 798–807. [Google Scholar] [CrossRef]
- Feron, O.; Balligand, J.L. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc. Res. 2006, 69, 788–797. [Google Scholar] [CrossRef]
- Pound, E.M.; Kang, J.X.; Leaf, A. Partitioning of polyunsaturated fatty acids, which prevent cardiac arrhythmias, into phospholipid cell membranes. J. Lipid Res. 2001, 42, 346–351. [Google Scholar]
- Ganotakis, E.S.; Mikhailidis, D.P.; Vardas, P.E. Atrial fibrillation, inflammation and statins. Helle. J. Cardiol. 2006, 47, 51–53. [Google Scholar]
- Young-Xu, Y.; Jabbour, S.; Goldberg, R.; Blatt, C.M.; Graboys, T.; Bilchik, B.; Ravid, S. Usefulness of statin drugs in protecting against atrial fibrillation in patients with coronary artery disease. Am. J. Cardiol. 2003, 92, 1379–1383. [Google Scholar] [CrossRef]
- Dernellis, J.; Panaretou, M. C-reactive protein and paroxysmal atrial fibrillation: Evidence of the implication of an inflammatory process in paroxysmal atrial fibrillation. Acta Cardiol. 2001, 56, 375–380. [Google Scholar] [CrossRef]
- Sarr, F.S.; André, C.; Guillaume, Y.C. Statins (HMG-coenzyme A reductase inhibitors)-biomimetic membrane binding mechanism investigated by molecular chromatography. J. Chromatogr. B 2008, 868, 20–27. [Google Scholar] [CrossRef]
- Davidson, M.H.; Toth, P.P. Comparative effects of lipid-lowering therapies. Prog. Cardiovasc. Dis. 2004, 47, 73–104. [Google Scholar] [CrossRef]
- Antoons, G.; Oros, A.; Bito, V.; Sipido, K.R.; Vos, M.A. Cellular basis for triggered ventricular arrhythmias that occur in the setting of compensated hypertrophy and heart failure: Considerations for diagnosis and treatment. J. Electrocardiol. 2007, 40, S8–S14. [Google Scholar]
- Mørk, H.K.; Sjaastad, I.; Sande, J.B.; Periasamy, M.; Sejersted, O.M.; Louch, W.E. Increased cardiomyocyte function and Ca2+ transients in mice during early congestive heart failure. J. Mol. Cell. Cardiol. 2007, 43, 177–186. [Google Scholar] [CrossRef]
- Del Monte, F.; Lebeche, D.; Guerrero, J.L.; Tsuji, T.; Doye, A.A.; Gwathmey, J.K.; Hajjar, R.J. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. PNAS 2004, 101, 5622–5627. [Google Scholar] [CrossRef]
- Satoh, M.; Ogita, H.; Takeshita, K.; Mukai, Y.; Kwiatkowski, D.J.; Liao, J.K. Requirement of Rac1 in the development of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 7432–7437. [Google Scholar] [CrossRef]
- Adam, O.; Frost, G.; Custodis, F.; Sussman, M.A.; Schäfers, H.J.; Böhm, M.; Laufs, U. Role of Rac1 GTPase activation in atrial fibrillation. J. Am. Coll. Cardiol. 2007, 50, 359–367. [Google Scholar] [CrossRef]
- Cha, T.J.; Ehrlich, J.R.; Zhang, L.; Shi, Y.F.; Tardif, J.C.; Leung, T.K.; Nattel, S. Dissociation between ionic remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation 2004, 109, 412–418. [Google Scholar] [CrossRef]
- Patel, R.; Nagueh, S.F.; Tsybouleva, N.; Abdellatif, M.; Lutucuta, S.; Kopelen, H.A.; Quinones, M.A.; Zoghbi, W.A.; Entman, M.L.; Roberts, R.; et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2001, 104, 317–324. [Google Scholar] [CrossRef]
- Stmbler, B.S.; Fenelon, G.; Shepard, R.K.; Clemo, H.F.; Guiraudon, C.M. Characterization of sustained atrial tachycardia in dogs with rapid ventricular pacing-induced heart failure. J. Cardiovasc. Electrophysiol. 2003, 14, 499–507. [Google Scholar] [CrossRef]
- Shiroshita-Takeshita, A.; Schram, G.; Lavoie, J.; Nattel, S. Effect of simvastatin and antioxidant vitamins on atrial fibrillation promotion by atrial-tachycardia remodeling in dogs. Circulation 2004, 110, 2313–2319. [Google Scholar] [CrossRef]
- Miyano, K.; Sumimoto, H. Role of the small GTPase Rac in p22phox-dependent NADPH oxidases. Biochimie 2007, 89, 1133–1144. [Google Scholar] [CrossRef]
- Mizuno, T.; Kaibuchi, K.; Ando, S.; Musha, T.; Hiraoka, K.; Takaishi, K.; Asada, M.; Nunoi, H.; Matsuda, I.; Takai, Y. Regulation of the superoxide-generating NADPH oxidase by a small GTP-binding protein and its stimulatory and inhibitory GDP/GTP exchange proteins. J. Biol. Chem. 1992, 267, 10215–10218. [Google Scholar]
- Cangemi, R.; Celestini, A.; Calvieri, C.; Carnevale, R.; Pastori, D.; Nocella, C.; Vicario, T.; Pignatelli, P.; Violi, F. Different behaviour of NOX2 activation in patients with paroxysmal/persistent or permanent atrial fibrillation. Heart 2012, 98, 1063–1066. [Google Scholar] [CrossRef]
- Laufs, U.; Kilter, H.; Konkol, C.; Wassmann, S.; Böhm, M.; Nickenig, G. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc. Res. 2002, 3, 911–920. [Google Scholar]
- Svetlana, N.R.; Raja, J.; Keshav, N.; Charalambos, A.; Sander, V.; Keith, M.C.; Nicholas, J.A.; Ulrich, S.; Barbara, C. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: Implications for the antiarrhythmic effect of statins. Circulation 2011, 124, 1107–1117. [Google Scholar] [CrossRef]
- Fukuchi, M.; Hussain, S.N.; Giaid, A. Heterogeneous expression and activity of endothelial and inducible nitric oxide synthases in end-stage human heart failure: their relation to lesion site and beta-adrenergic receptor therapy. Circulation 1998, 98, 132–139. [Google Scholar] [CrossRef]
- Fischmeister, R.; Castro, L.; Abi-Gerges, A.; Rochais, F.; Vandecasteele, G. Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comp. Biochem. Physiol. A 2005, 142, 136–143. [Google Scholar] [CrossRef]
- Massion, P.B.; Feron, O.; Dessy, C.; Balligand, J.L. Nitric oxide and cardiac function: Ten years after, and continuing. Circ. Res. 2003, 93, 388–398. [Google Scholar] [CrossRef]
- Damy, T.; Ratajczak, P.; Shah, A.M.; Camors, E.; Marty, I.; Hasenfuss, G.; Marotte, F.; Samuel, J.L.; Heymes, C. Increased neuronal nitric oxide synthase-derived NO production in the failing human heart. Lancet 2004, 363, 1365–1367. [Google Scholar] [CrossRef]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial NOX2 containing NADPH oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef]
- Dudley, S.C., Jr.; Hoch, N.E.; McCann, L.A., Honeycutt; Diamandopoulos, L.; Fukai, T.; Harrison, D.G.; Dikalov, S.I.; Langberg, J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. [Google Scholar] [CrossRef]
- Kim, H.S.; No, C.W.; Goo, S.H.; Cha, T.J. An angiotensin receptor blocker prevents arrhythmogenic left atrial remodeling in a rat post myocardial infarction induced heart failure model. J. Korean Med. Sci. 2013, 28, 700–708. [Google Scholar] [CrossRef]
- Pogwizd, S.M.; Schlotthauer, K.; Li, L.; Yuan, W.; Bers, D.M. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsivenes. Circ. Res. 2001, 88, 1159–1167. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Festing, M.F.; Altman, D.G. Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR J. 2002, 43, 244–228. [Google Scholar] [CrossRef]
- Violi, F.; Loffredo, L. Thromboembolism or atherothromboembolism in atrial fibrillation? Circ. Arrhythm. Electrophysiol. 2012, 5, 1053–1055. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cho, K.-I.; Koo, S.-H.; Cha, T.-J.; Heo, J.-H.; Kim, H.-S.; Jo, G.-B.; Lee, J.-W. Simvastatin Attenuates the Oxidative Stress, Endothelial Thrombogenicity and the Inducibility of Atrial Fibrillation in a Rat Model of Ischemic Heart Failure. Int. J. Mol. Sci. 2014, 15, 14803-14818. https://doi.org/10.3390/ijms150814803
Cho K-I, Koo S-H, Cha T-J, Heo J-H, Kim H-S, Jo G-B, Lee J-W. Simvastatin Attenuates the Oxidative Stress, Endothelial Thrombogenicity and the Inducibility of Atrial Fibrillation in a Rat Model of Ischemic Heart Failure. International Journal of Molecular Sciences. 2014; 15(8):14803-14818. https://doi.org/10.3390/ijms150814803
Chicago/Turabian StyleCho, Kyoung-Im, Sang-Ho Koo, Tae-Joon Cha, Jung-Ho Heo, Hyun-Su Kim, Gee-Bum Jo, and Jae-Woo Lee. 2014. "Simvastatin Attenuates the Oxidative Stress, Endothelial Thrombogenicity and the Inducibility of Atrial Fibrillation in a Rat Model of Ischemic Heart Failure" International Journal of Molecular Sciences 15, no. 8: 14803-14818. https://doi.org/10.3390/ijms150814803