What Controls DNA Looping?

Abstract
:
1. Introduction

2. Results and Discussion
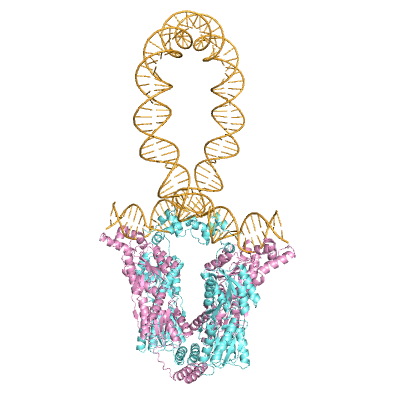
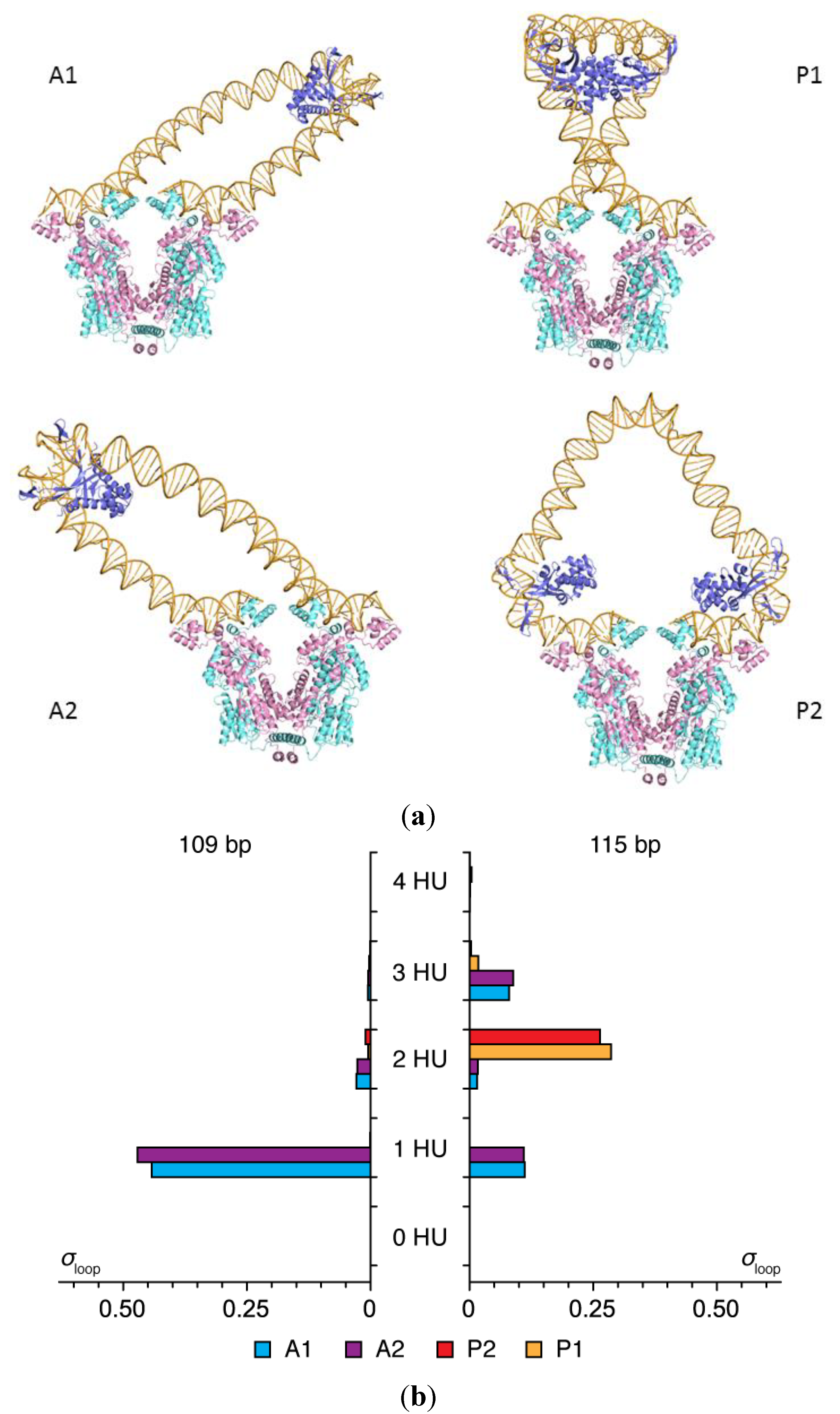
2.1. Contributions of HU to DNA Looping

2.2. Effects of the Lac Repressor on DNA Loops

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Loop | Ebend (kBT) | Etwist (kBT) | σloop (%) | Jloop (%) |
|---|---|---|---|---|
| A1 | 18.7 | 8.6 | 28.3 | 39.8 |
| A2 | 18.8 | 8.4 | 30.3 | 29.7 |
| P1 | 22.9 | 4.0 | 41.4 | 28.2 |
| P2 | 31.4 | 32.9 | 0 | 2.3 |

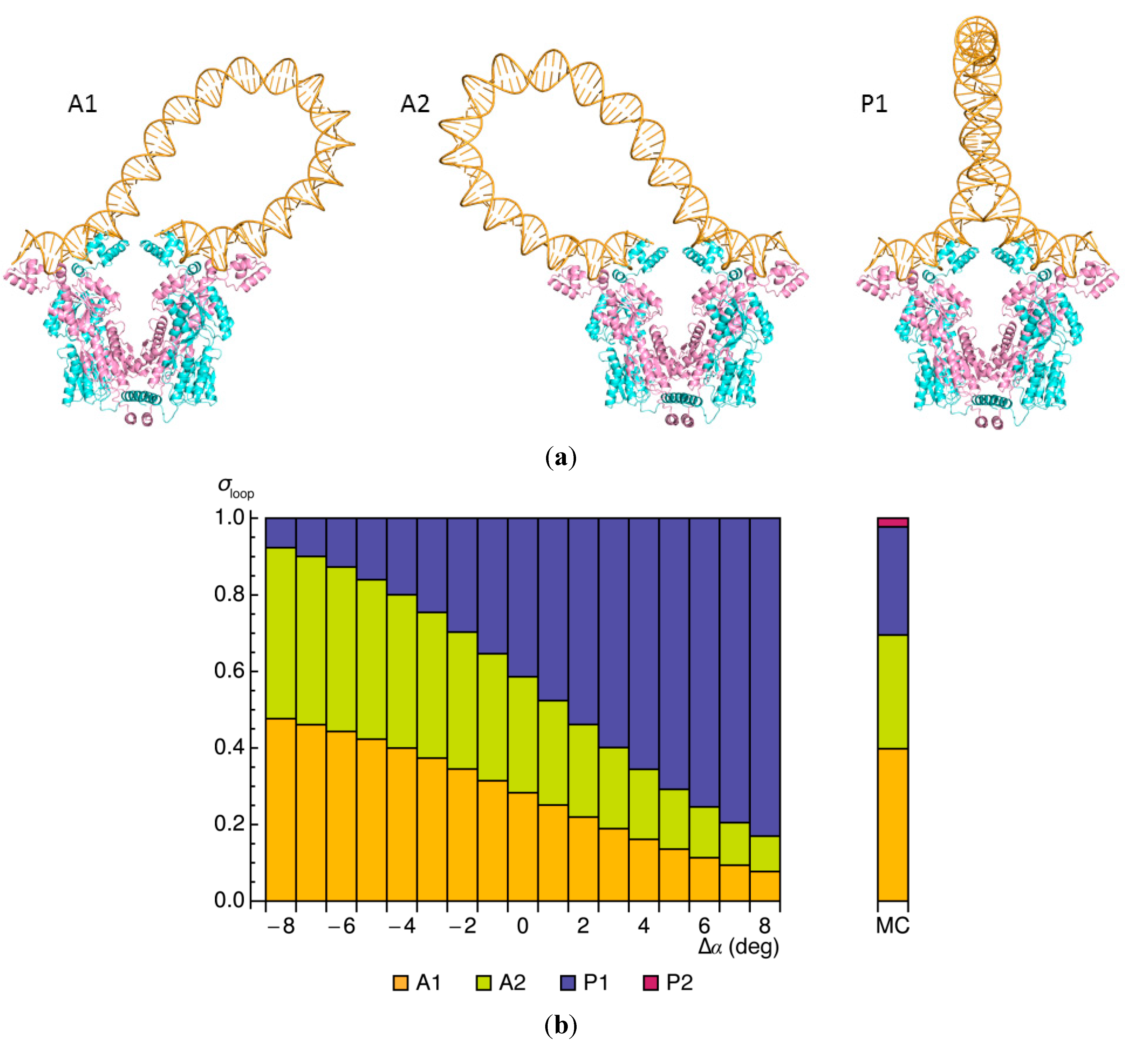
2.3. Influences of Sequence and Environment on Looped Structures


2.4. Effects of Operator Structure on Looping Propensities
 ). Note the enhancement in looping with the natural operators.
). Note the enhancement in looping with the natural operators.
). Note the enhancement in looping with the natural operators.
). Note the enhancement in looping with the natural operators.
3. Experimental Section
3.1. Protein-Decorated DNA Model
3.2. Looping Simulations
3.3. Loop Optimization
3.4. Loop Characterization and Manipulation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Müller-Hill, B. The lac Operon; Walter de Gruyter: Berlin, Germany, 1996. [Google Scholar]
- Koh, J.; Shkel, I.; Saecker, R.M.; Record, M.T., Jr. Nonspecific DNA binding and bending by HUαβ: Interfaces of the three binding modes characterized by salt-dependent thermodynamics. J. Mol. Biol. 2011, 410, 241–267. [Google Scholar] [CrossRef]
- Swinger, K.K.; Lemberg, K.M.; Zhang, Y.; Rice, P.A. Flexible DNA bending in HU–DNA cocrystal structures. EMBO J. 2003, 22, 3749–3760. [Google Scholar] [CrossRef]
- Krämer, H.; Niemöller, M.; Amouyal, M.; Revêt, B.; von Wilcken-Bergmann, B.; Müller-Hill, B. Lac repressor forms loops with linear DNA carrying two suitably spaced lac operators. EMBO J. 1987, 6, 1481–1491. [Google Scholar]
- Bond, L.M.; Peters, J.P.; Becker, N.A.; Kahn, J.D.; Maher, L.J., 3rd. Gene repression by minimal lac loops in vivo. Nucleic Acids Res. 2010, 38, 8071–8082. [Google Scholar]
- Lewis, M.; Chang, G.; Horton, N.C.; Kercher, M.A.; Pace, H.C.; Schumacher, M.A.; Brennan, R.G.; Lu, P. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science 1996, 271, 1247–1254. [Google Scholar]
- Ruben, G.C.; Roos, T.B. Conformation of Lac repressor tetramer in solution, bound and unbound to operator DNA. Microsc. Res. Tech. 1997, 36, 400–416. [Google Scholar] [CrossRef]
- Taraban, M.; Zhan, H.; Whitten, A.E.; Langley, D.B.; Matthews, K.S.; Swint-Kruse, L.; Trewhella, J. Ligand-induced conformational changes and conformational dynamics in the solution structure of the lactose repressor protein. J. Mol. Biol. 2008, 376, 466–481. [Google Scholar] [CrossRef]
- Colasanti, A.V.; Grosner, M.A.; Perez, P.J.; Clauvelin, N.; Lu, X.-J.; Olson, W.K. Weak operator binding enhances simulated Lac repressor-mediated DNA looping. Biopolymers 2013, 99, 1070–1081. [Google Scholar]
- Villa, E.; Balaeff, A.; Schulten, K. Structural dynamics of the Lac repressor–DNA complex revealed by a multiscale simulation. Proc. Natl. Acad. Sci. USA 2005, 102, 6783–6788. [Google Scholar] [CrossRef]
- Olson, W.K.; Gorin, A.A.; Lu, X.-J.; Hock, L.M.; Zhurkin, V.B. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA 1998, 95, 11163–11168. [Google Scholar]
- Tolstorukov, M.Y.; Colasanti, A.V.; McCandlish, D.M.; Olson, W.K.; Zhurkin, V.B. A novel roll-and-slide mechanism of DNA folding in chromatin: Implications for nucleosome positioning. J. Mol. Biol. 2007, 371, 725–738. [Google Scholar] [CrossRef]
- Geanacopoulos, M.; Vasmatzis, G.; Zhurkin, V.B.; Adhya, S. Gal repressosome contains an antiparallel DNA loop. Nat. Struct. Biol. 2001, 8, 432–436. [Google Scholar] [CrossRef]
- Becker, N.A.; Kahn, J.D.; Maher, L.J., 3rd. Bacterial repression loops require enhanced DNA flexibility. J. Mol. Biol. 2005, 349, 716–730. [Google Scholar] [CrossRef]
- Becker, N.A.; Kahn, J.D.; Maher, L.J., 3rd. Effects of nucleoid proteins on DNA repression loop formation in Escherichia coli. Nucleic Acids Res. 2007, 35, 3988–4000. [Google Scholar] [CrossRef]
- Han, L.; Garcia, H.G.; Blumberg, S.; Towles, K.B.; Beausang, J.F.; Nelson, P.C.; Phillips, R. Concentration and length dependence of DNA looping in transcriptional regulation. PLoS One 2009, 4, e5621. [Google Scholar]
- Johnson, S.; Lindén, M.; Phillips, R. Sequence dependence of transcription factor-mediated DNA looping. Nucleic Acids Res. 2012, 40, 7728–7738. [Google Scholar] [CrossRef]
- Ali Azam, T.; Iwata, A.; Nishimura, A.; Ueda, S.; Ishihama, A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J. Bacteriol. 1999, 181, 6361–6370. [Google Scholar]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef]
- Boedicker, J.Q.; Garcia, H.G.; Johnson, S.; Phillips, R. DNA sequence-dependent mechanics and protein-assisted bending in repressor-mediated loop formation. Phys. Biol. 2013, 10, 066005. [Google Scholar] [CrossRef]
- Johnson, S. DNA Mechanics and Transcriptional Regulation in the E. coli lac Operon. Ph.D. Thesis, California Institute of Technology, Pasadena, CA, USA, 26 December 2012. [Google Scholar]
- PyMOL. Available online: http://www.pymol.org (accessed on 27 August 2014).
- Czapla, L.; Grosner, M.A.; Swigon, D.; Olson, W.K. Interplay of protein and DNA structure revealed in simulations of the lac operon. PLoS One 2013, 8, e56548. [Google Scholar]
- Swigon, D.; Coleman, B.D.; Olson, W.K. Modeling the Lac repressor-operator assembly: The influence of DNA looping on Lac repressor conformation. Proc. Natl. Acad. Sci. USA 2006, 103, 9879–9884. [Google Scholar] [CrossRef]
- Liu, J.S. Monte Carlo Strategies in Scientific Computing; Springer-Verlag: New York, NY, USA, 2001. [Google Scholar]
- White, J.H.; Bauer, W.R. Calculation of the twist and the writhe for representative models of DNA. J. Mol. Biol. 1986, 189, 329–341. [Google Scholar] [CrossRef]
- Depew, R.E.; Wang, J.C. Conformational fluctuations of DNA helix. Proc. Natl. Acad. Sci. USA 1975, 72, 4275–4279. [Google Scholar] [CrossRef]
- Anderson, P.; Bauer, W. Supercoiling in closed circular DNA: Dependence upon ion type and concentration. Biochemistry 1978, 17, 594–601. [Google Scholar] [CrossRef]
- Love, A.E.H. A Treatise on the Mathematical Theory of Elasticity, 4th ed.; Dover Publications: Mineola, NY, USA, 1944. [Google Scholar]
- Spronk, C.A.; Bonvin, A.M.; Radha, P.K.; Melacini, G.; Boelens, R.; Kaptein, R. The solution structure of Lac repressor headpiece 62 complexed to a symmetrical lac operator. Structure 1999, 7, 1483–1492. [Google Scholar] [CrossRef]
- Romanuka, J.; Folkers, G.E.; Biris, N.; Tishchenko, E.; Wienk, H.; Bonvin, A.M.J.J.; Kaptein, R.; Boelens, R. Specificity and affinity of Lac repressor for the auxiliary operators O2 and O3 are explained by the structures of their protein–DNA complexes. J. Mol. Biol. 2009, 390, 478–489. [Google Scholar] [CrossRef]
- Lu, X.J.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef]
- Lu, X.-J.; Olson, W.K. 3DNA: A versatile, integrated software system for the analysis, rebuilding, and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008, 3, 1213–1227. [Google Scholar] [CrossRef]
- Czapla, L.; Swigon, D.; Olson, W.K. Sequence-dependent effects in the cyclization of short DNA. J. Chem. Theor. Comp. 2006, 2, 685–695. [Google Scholar] [CrossRef]
- Horowitz, D.S.; Wang, J.C. Torsional rigidity of DNA and length dependence of the free energy of DNA supercoiling. J. Mol. Biol. 1984, 173, 75–91. [Google Scholar] [CrossRef]
- Heath, P.J.; Clendenning, J.B.; Fujimoto, B.S.; Schurr, J.M. Effect of bending strain on the torsion elastic constant of DNA. J. Mol. Biol. 1996, 260, 718–730. [Google Scholar] [CrossRef]
- Czapla, L.; Swigon, D.; Olson, W.K. Effects of the nucleoid protein HU on the structure, flexibility, and ring-closure properties of DNA deduced from Monte Carlo simulations. J. Mol. Biol. 2008, 382, 353–370. [Google Scholar] [CrossRef]
- Bell, C.E.; Lewis, M. A closer view of the conformation of the Lac repressor bound to operator. Nat. Struct. Biol. 2000, 7, 209–214. [Google Scholar] [CrossRef]
- Bell, C.E.; Lewis, M. Crystallographic analysis of Lac repressor bound to natural operator O1. J. Mol. Biol. 2001, 312, 921–926. [Google Scholar] [CrossRef]
- Jacobson, H.; Stockmayer, W.H. Intramolecular reaction in polycondensations. I. The theory of linear systems. J. Chem. Phys. 1950, 18, 1600–1606. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes in C, 2nd ed.; Cambridge University Press: New York, NY, USA, 1992; pp. 288–290. [Google Scholar]
- Alexandrowicz, Z. Monte Carlo of chains with excluded volume: A way to evade sample attrition. J. Chem. Phys. 1969, 51, 561–565. [Google Scholar] [CrossRef]
- Clauvelin, N.; Olson, W.K. The Synergy between Protein Positioning and DNA Elasticity: Energy Minimization of Protein-Decorated DNA Minicircles. 2014. Available online: http://arxiv.org/abs/1405.7638 (accessed on 29 May 2014).
- Dickerson, R.E.; Bansal, M.; Calladine, C.R.; Diekmann, S.; Hunter, W.N.; Kennard, O.; von Kitzing, E.; Lavery, R.; Nelson, H.C.M.; Olson, W.K.; et al. Definitions and nomenclature of nucleic acid structure parameters. Nucleic Acids Res. 1989, 17, 1797–1803. [Google Scholar] [CrossRef]
- El Hassan, M.A.; Calladine, C.R. The assessment of the geometry of dinucleotide steps in double-helical DNA: A new local calculation scheme. J. Mol. Biol. 1995, 251, 648–664. [Google Scholar] [CrossRef]
- Clauvelin, N.; Tobias, I.; Olson, W.K. Characterization of the geometry and topology of DNA pictured as a discrete collection of atoms. J. Chem. Theor. Comp. 2012, 8, 1092–1107. [Google Scholar] [CrossRef]
- Britton, L.; Olson, W.K.; Tobias, I. Two perspectives on the twist of DNA. J. Chem. Phys. 2009, 131, 245101. [Google Scholar] [CrossRef]
- Colasanti, A.V.; Lu, X.J.; Olson, W.K. Analyzing and building nucleic acid structures with 3DNA. J. Vis. Exp. 2013, 74, e4401. [Google Scholar]
- Zhang, Y.; McEwen, A.E.; Crothers, D.M.; Levene, S.D. Analysis of in vivo LacR-mediated gene repression based on the mechanics of DNA looping. PLoS One 2006, 1, e136. [Google Scholar] [CrossRef]
- Saiz, L.; Vilar, J.M. Multilevel deconstruction of the in vivo behavior of looped DNA–protein complexes. PLoS One 2007, 2, e355. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Perez, P.J.; Clauvelin, N.; Grosner, M.A.; Colasanti, A.V.; Olson, W.K. What Controls DNA Looping? Int. J. Mol. Sci. 2014, 15, 15090-15108. https://doi.org/10.3390/ijms150915090
Perez PJ, Clauvelin N, Grosner MA, Colasanti AV, Olson WK. What Controls DNA Looping? International Journal of Molecular Sciences. 2014; 15(9):15090-15108. https://doi.org/10.3390/ijms150915090
Chicago/Turabian StylePerez, Pamela J., Nicolas Clauvelin, Michael A. Grosner, Andrew V. Colasanti, and Wilma K. Olson. 2014. "What Controls DNA Looping?" International Journal of Molecular Sciences 15, no. 9: 15090-15108. https://doi.org/10.3390/ijms150915090