Mechanical Forces Induce Changes in VEGF and VEGFR-1/sFlt-1 Expression in Human Chondrocytes


 and
and 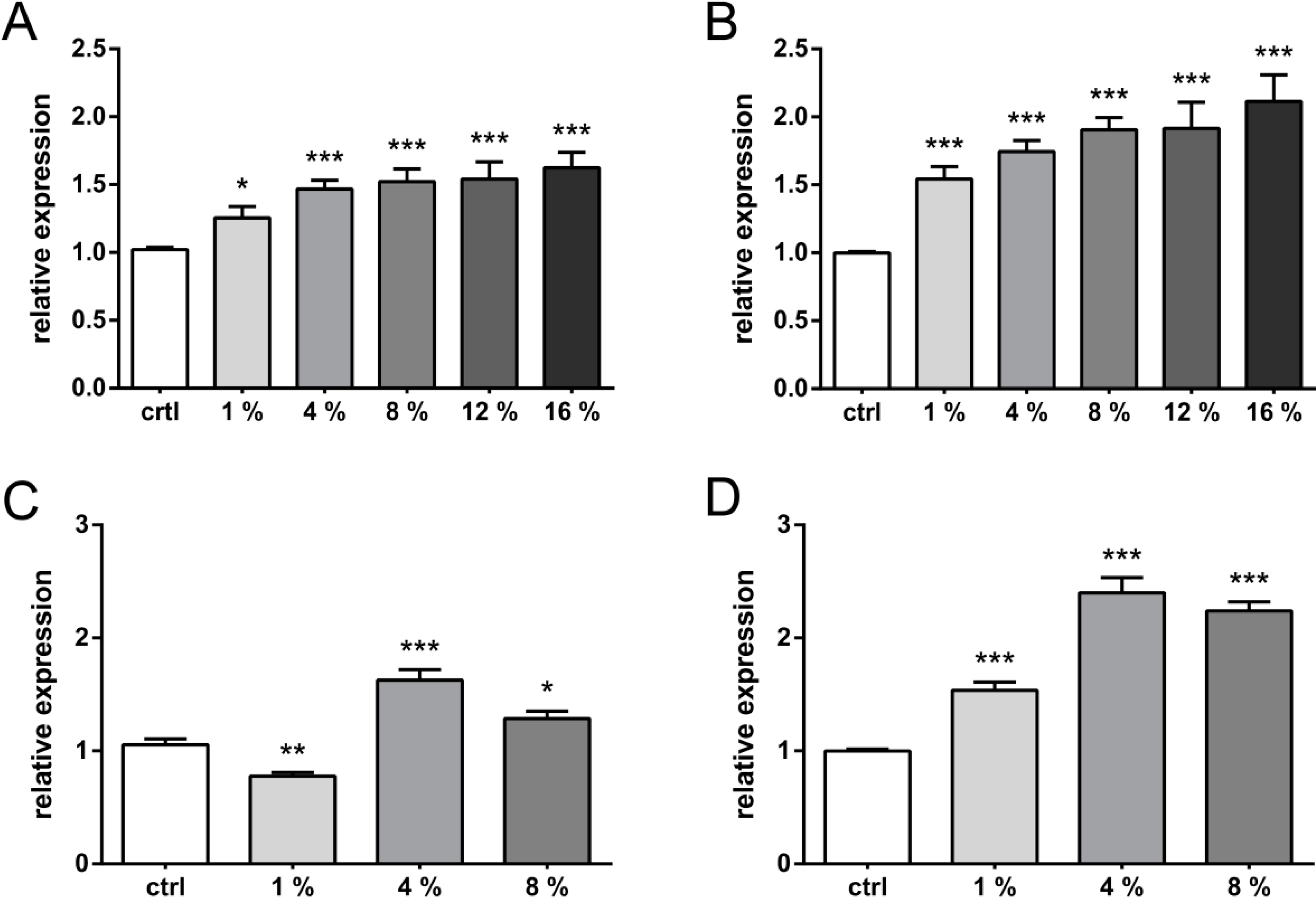{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
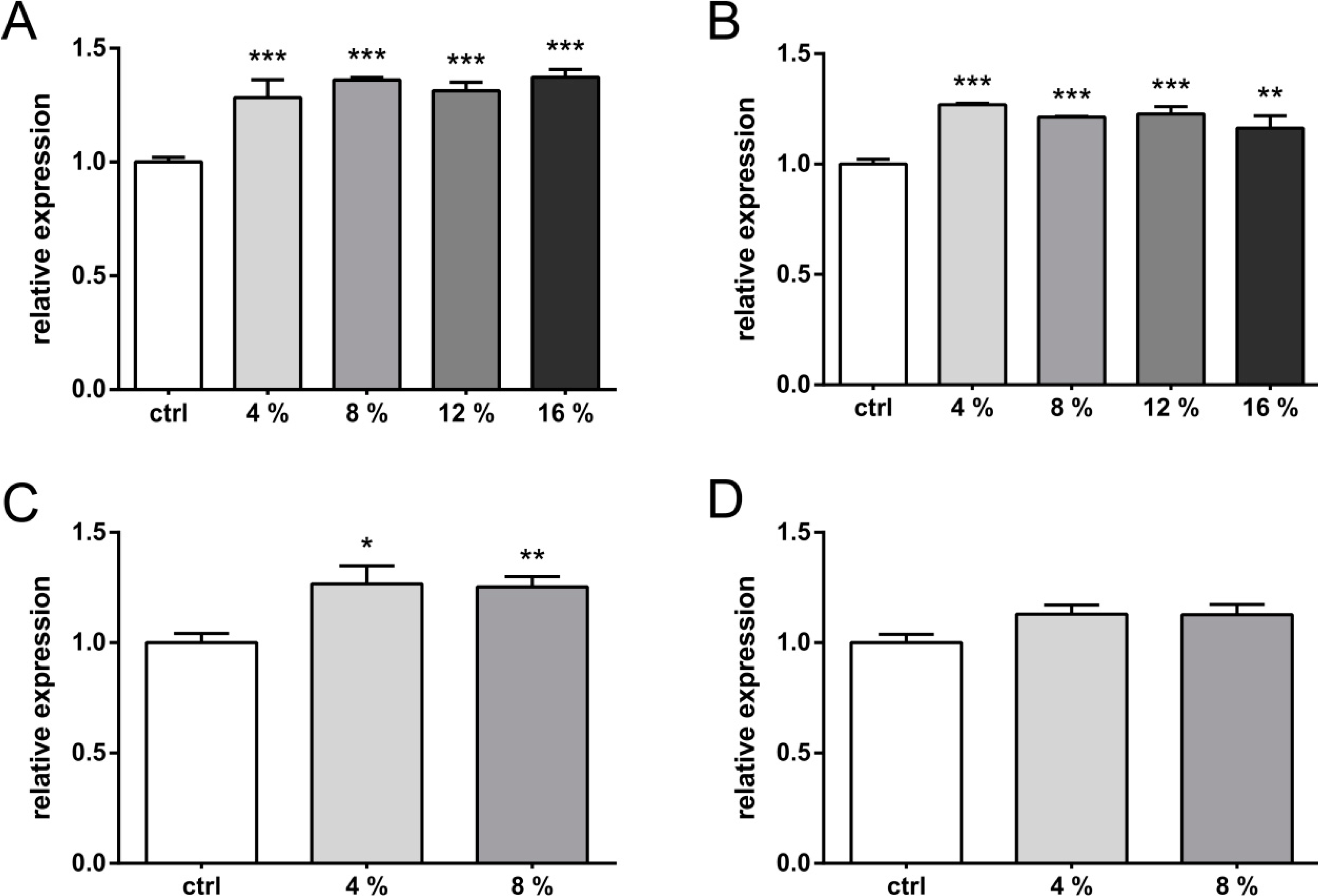
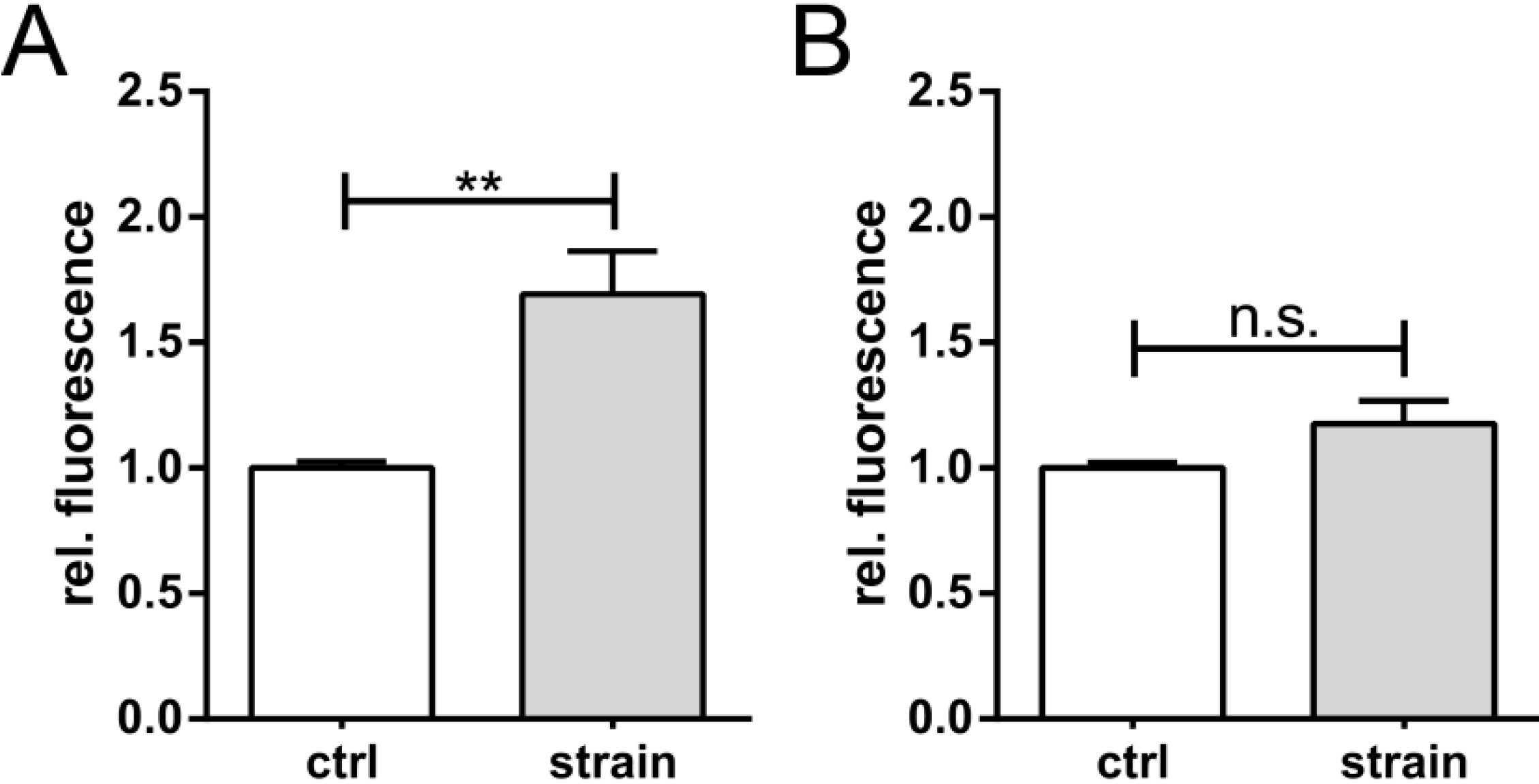
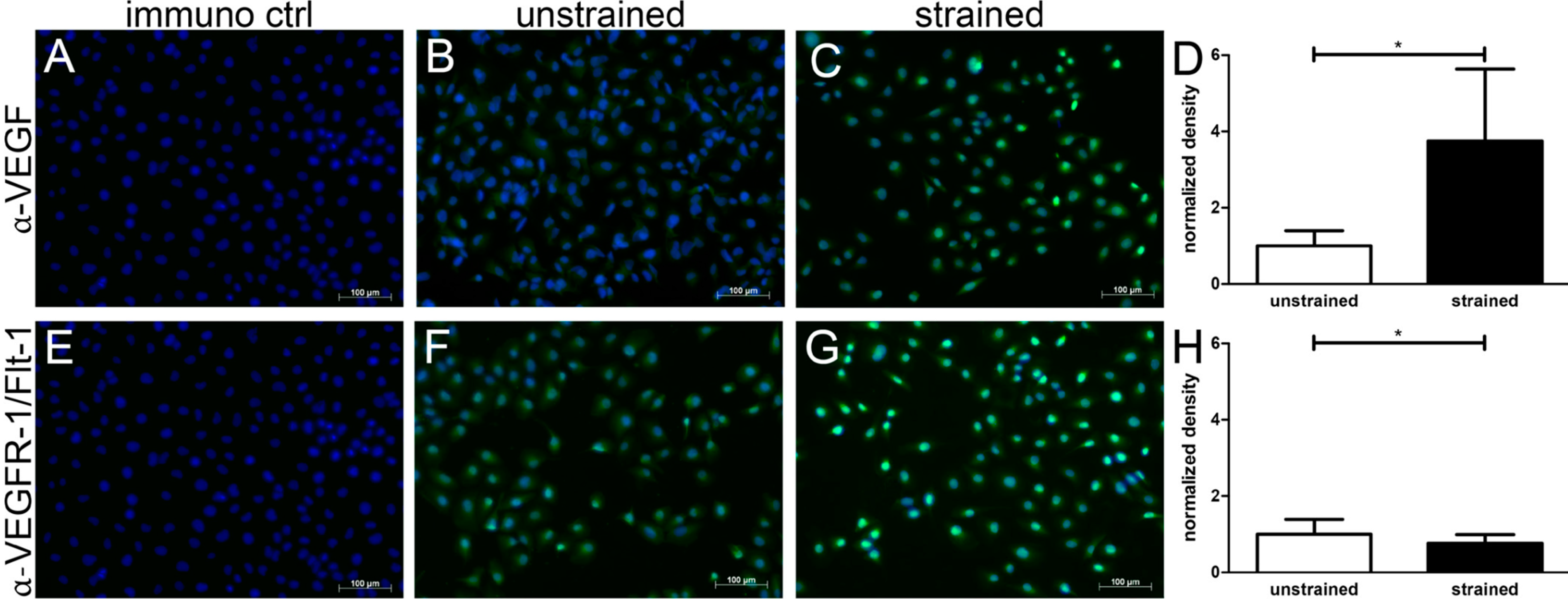
2. Results and Discussion
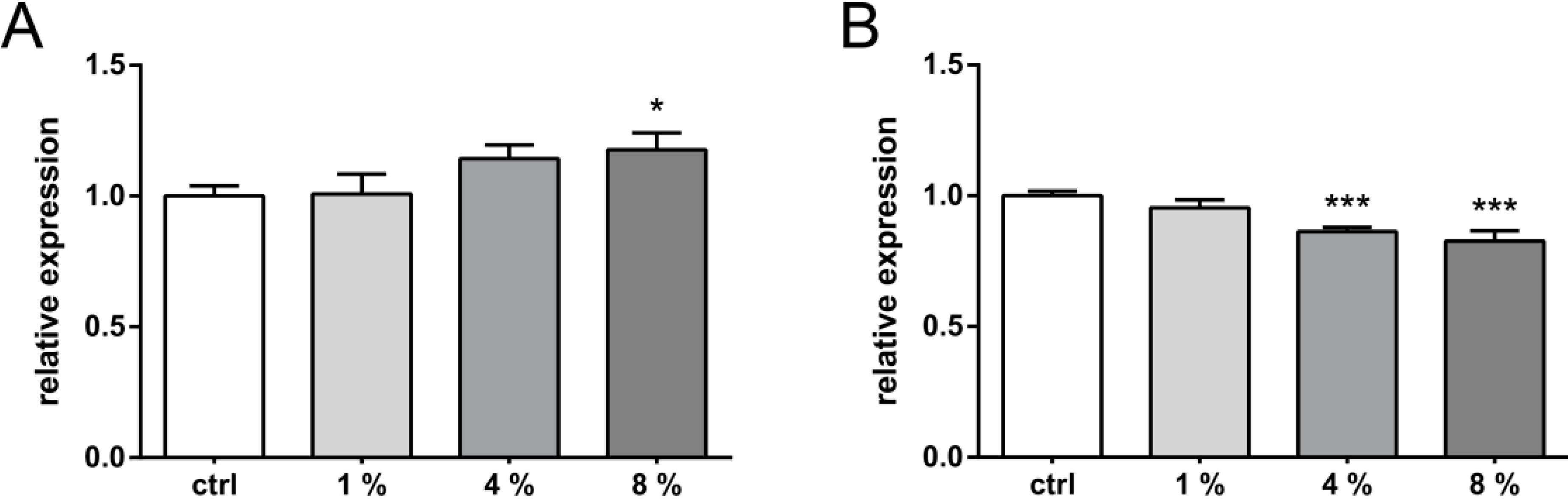
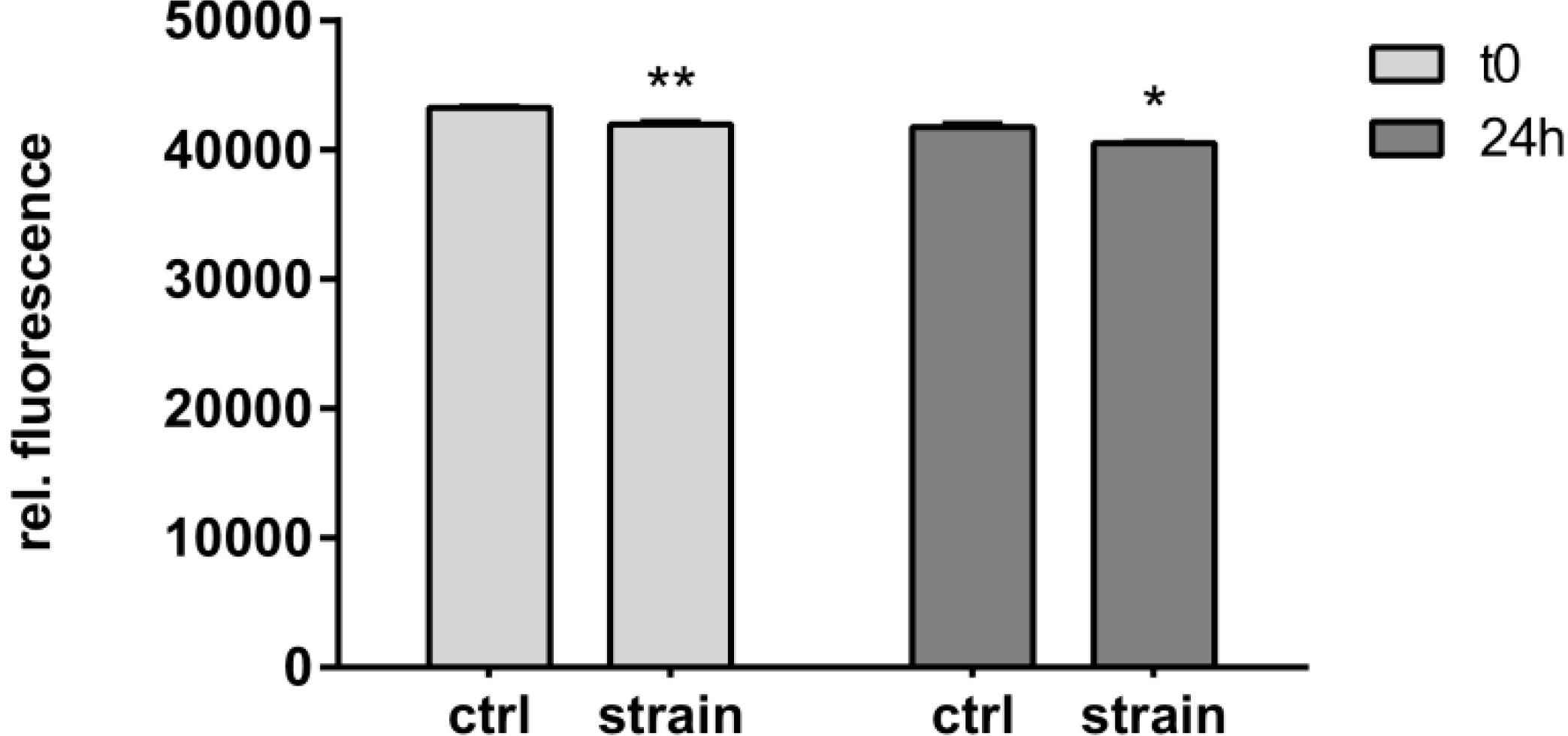
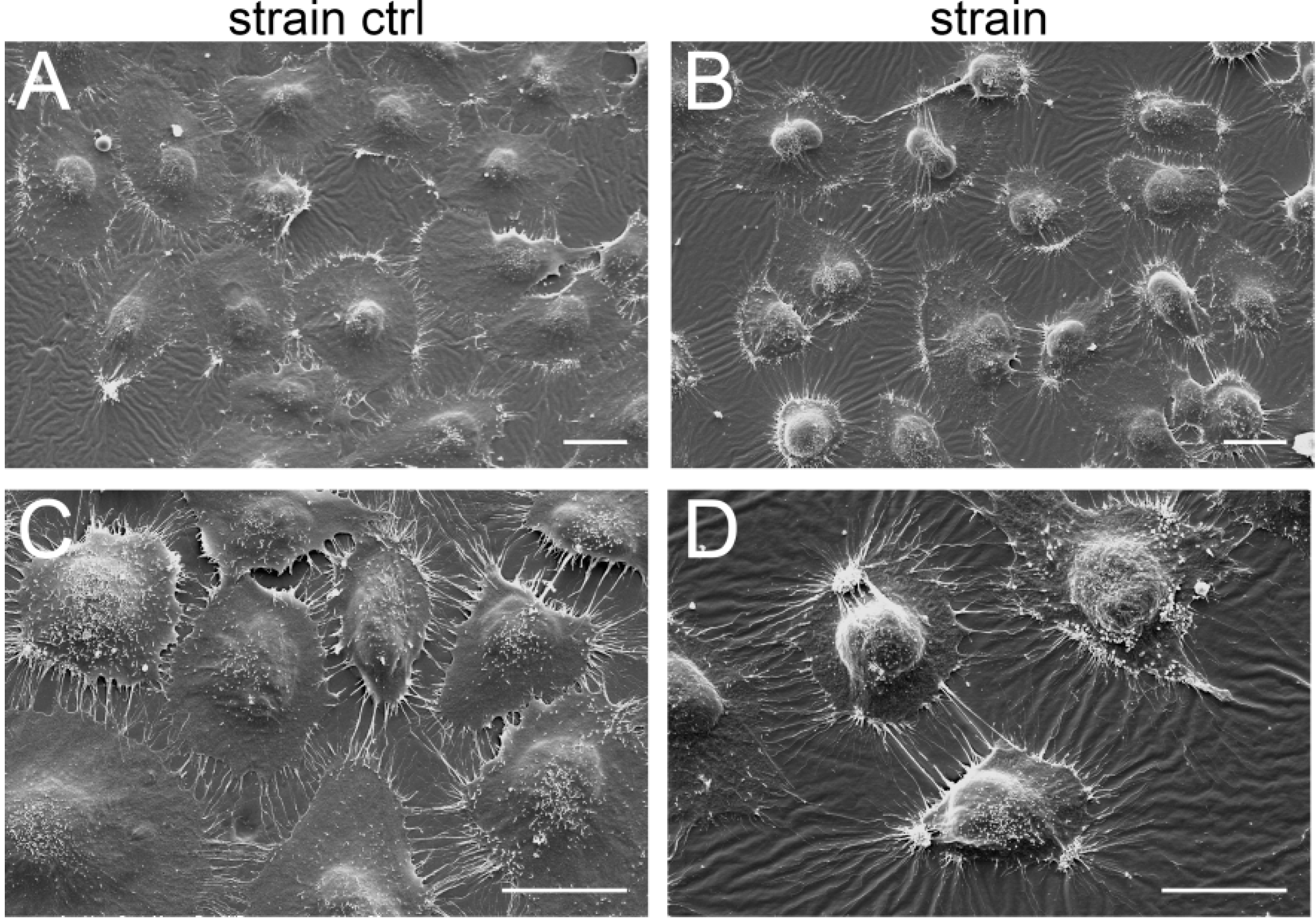
2.1. Results







2.2. Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Mechanical Stimulation
3.3. Biochemical Analyses
3.4. Cell Viability Assay
3.5. Transient Transfections and Dual Luciferase Assays
3.6. Immunofluoresence
3.7. Scanning Electron Microscopy (SEM)
3.8. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guilak, F.; Ratcliffe, A.; Mow, V.C. Chondrocyte deformation and local tissue strain in articular cartilage: A confocal microscopy study. J. Orthop. Res. 1995, 13, 410–421. [Google Scholar] [CrossRef]
- Leong, D.J.; Li, Y.H.; Gu, X.I.; Sun, L.; Zhou, Z.; Nasser, P.; Laudier, D.M.; Iqbal, J.; Majeska, R.J.; Schaffler, M.B.; et al. Physiological loading of joints prevents cartilage degradation through cited2. FASEB J. 2011, 25, 182–191. [Google Scholar] [CrossRef]
- Holmvall, K.; Camper, L.; Johansson, S.; Kimura, J.H.; Lundgren-Akerlund, E. Chondrocyte and chondrosarcoma cell integrins with affinity for collagen type II and their response to mechanical stress. Exp. Cell Res. 1995, 221, 496–503. [Google Scholar] [CrossRef]
- Akagi, M.; Nishimura, S.; Yoshida, K.; Kakinuma, T.; Sawamura, T.; Munakata, H.; Hamanishi, C. Cyclic tensile stretch load and oxidized low density lipoprotein synergistically induce lectin-like oxidized LDL receptor-1 in cultured bovine chondrocytes, resulting in decreased cell viability and proteoglycan synthesis. J. Orthop. Res. 2006, 24, 1782–1790. [Google Scholar] [CrossRef]
- Millward-Sadler, S.J.; Wright, M.O.; Davies, L.W.; Nuki, G.; Salter, D.M. Mechanotransduction via integrins and interleukin-4 results in altered aggrecan and matrix metalloproteinase 3 gene expression in normal, but not osteoarthritic, human articular chondrocytes. Arthritis Rheumatol. 2000, 43, 2091–2099. [Google Scholar] [CrossRef]
- Das, R.H.; Jahr, H.; Verhaar, J.A.; van der Linden, J.C.; van Osch, G.J.; Weinans, H. In vitro expansion affects the response of chondrocytes to mechanical stimulation. Osteoarthr. Cartil. 2008, 16, 385–391. [Google Scholar] [CrossRef]
- Loeser, R.F. Molecular mechanisms of cartilage destruction: Mechanics, inflammatory mediators, and aging collide. Arthritis Rheumatol. 2006, 54, 1357–1360. [Google Scholar] [CrossRef]
- Tanaka, S.; Hamanishi, C.; Kikuchi, H.; Fukuda, K. Factors related to degradation of articular cartilage in osteoarthritis: A review. Semin. Arthritis Rheum. 1998, 27, 392–399. [Google Scholar] [CrossRef]
- Behrens, F.; Kraft, E.L.; Oegema, T.R., Jr. Biochemical changes in articular cartilage after joint immobilization by casting or external fixation. J. Orthop. Res. 1989, 7, 335–343. [Google Scholar] [CrossRef]
- Slowman, S.D.; Brandt, K.D. Composition and glycosaminoglycan metabolism of articular cartilage from habitually loaded and habitually unloaded sites. Arthritis Rheumatol. 1986, 29, 88–94. [Google Scholar] [CrossRef]
- Hatabu, T.; Kawazu, S.; Kojima, S.; Sato, K.; Singhasivanon, P.; Looareesuwan, S.; Kano, S. In vitro susceptibility and genetic variations for chloroquine and mefloquine in plasmodium falciparum isolates from thai-myanmar border. Southeast Asian J. Trop. Med. Public Health 2005, 36, 73–79. [Google Scholar]
- Davis, M.A.; Ettinger, W.H.; Neuhaus, J.M.; Cho, S.A.; Hauck, W.W. The association of knee injury and obesity with unilateral and bilateral osteoarthritis of the knee. Am. J. Epidemiol. 1989, 130, 278–288. [Google Scholar]
- Gelber, A.C.; Hochberg, M.C.; Mead, L.A.; Wang, N.Y.; Wigley, F.M.; Klag, M.J. Joint injury in young adults and risk for subsequent knee and hip osteoarthritis. Ann. Intern. Med. 2000, 133, 321–328. [Google Scholar] [CrossRef]
- Murphy, S.B.; Ganz, R.; Muller, M.E. The prognosis in untreated dysplasia of the hip. A study of radiographic factors that predict the outcome. J. Bone Jt. Surg. Am. Vol. 1995, 77, 985–989. [Google Scholar]
- Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of human knee menisci: Structure, composition, and function. Sports Health 2009, 4, 340–351. [Google Scholar]
- Pufe, T.; Petersen, W.; Tillmann, B.; Mentlein, R. The splice variants VEGF121 and VEGF189 of the angiogenic peptide vascular endothelial growth factor are expressed in osteoarthritic cartilage. Arthritis Rheumatol. 2001, 44, 1082–1088. [Google Scholar] [CrossRef]
- Pufe, T.; Kurz, B.; Petersen, W.; Varoga, D.; Mentlein, R.; Kulow, S.; Lemke, A.; Tillmann, B. The influence of biomechanical parameters on the expression of VEGF and endostatin in the bone and joint system. Ann. Anat. 2005, 187, 461–472. [Google Scholar] [CrossRef]
- Eisenstein, R.; Sorgente, N.; Soble, L.W.; Miller, A.; Kuettner, K.E. The resistance of certain tissues to invasion: Penetrability of explanted tissues by vascularized mesenchyme. Am. J. Pathol. 1973, 73, 765–774. [Google Scholar]
- Fenwick, S.A.; Gregg, P.J.; Rooney, P. Osteoarthritic cartilage loses its ability to remain avascular. Osteoarthr. Cartil. 1999, 7, 441–452. [Google Scholar] [CrossRef]
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef]
- Wong, M.; Siegrist, M.; Goodwin, K. Cyclic tensile strain and cyclic hydrostatic pressure differentially regulate expression of hypertrophic markers in primary chondrocytes. Bone 2003, 33, 685–693. [Google Scholar] [CrossRef]
- Pfander, D.; Kortje, D.; Zimmermann, R.; Weseloh, G.; Kirsch, T.; Gesslein, M.; Cramer, T.; Swoboda, B. Vascular endothelial growth factor in articular cartilage of healthy and osteoarthritic human knee joints. Ann. Rheum. Dis. 2001, 60, 1070–1073. [Google Scholar] [CrossRef]
- Enomoto, H.; Inoki, I.; Komiya, K.; Shiomi, T.; Ikeda, E.; Obata, K.; Matsumoto, H.; Toyama, Y.; Okada, Y. Vascular endothelial growth factor isoforms and their receptors are expressed in human osteoarthritic cartilage. Am. J. Pathol. 2003, 162, 171–181. [Google Scholar] [CrossRef]
- Shakibaei, M.; Schulze-Tanzil, G.; Mobasheri, A.; Beichler, T.; Dressler, J.; Schwab, W. Expression of the vegf receptor-3 in osteoarthritic chondrocytes: Stimulation by interleukin-1β and association with β1-integrins. Histochem. Cell Biol. 2003, 120, 235–241. [Google Scholar]
- Pufe, T.; Harde, V.; Petersen, W.; Goldring, M.B.; Tillmann, B.; Mentlein, R. Vascular endothelial growth factor (VEGF) induces matrix metalloproteinase expression in immortalized chondrocytes. J. Pathol. 2004, 202, 367–374. [Google Scholar] [CrossRef]
- Studer, D.; Millan, C.; Ozturk, E.; Maniura-Weber, K.; Zenobi-Wong, M. Molecular and biophysical mechanisms regulating hypertrophic differentiation in chondrocytes and mesenchymal stem cells. Eur. Cells Mater. 2012, 24, 118–135. [Google Scholar]
- Murata, M.; Yudoh, K.; Masuko, K. The potential role of vascular endothelial growth factor (VEGF) in cartilage: How the angiogenic factor could be involved in the pathogenesis of osteoarthritis? Osteoarthr. Cartil. 2008, 16, 279–286. [Google Scholar] [CrossRef]
- Ladomery, M.R.; Harper, S.J.; Bates, D.O. Alternative splicing in angiogenesis: The vascular endothelial growth factor paradigm. Cancer Lett. 2007, 249, 133–142. [Google Scholar] [CrossRef]
- Dvorak, H.F. Angiogenesis: Update 2005. J. Thromb. Haemost. 2005, 3, 1835–1842. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. CellBiol. 1996, 16, 4604–4613. [Google Scholar]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.H. Different signal transduction properties of KDR and FLT1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar]
- Thomas, K.A. Vascular endothelial growth factor, a potent and selective angiogenic agent. J. Biol. Chem. 1996, 271, 603–606. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular endothelial growth factor: Basic science and clinical progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef]
- Herzog, W.; Clark, A.; Longino, D. Joint Mechanics in osteoarthritis. In Osteoarthritic Joint Pain; John Wiley & Sons, Ltd.: Chichester, UK, 2004; pp. 79–99. [Google Scholar]
- Felson, D.T. Osteoarthritis as a disease of mechanics. Osteoarthr. Cartil. 2013, 21, 10–15. [Google Scholar] [CrossRef]
- Ludin, A.; Sela, J.J.; Schroeder, A.; Samuni, Y.; Nitzan, D.W.; Amir, G. Injection of vascular endothelial growth factor into knee joints induces osteoarthritis in mice. Osteoarthr. Cartil. 2013, 21, 491–497. [Google Scholar] [CrossRef]
- Mapp, P.I.; Walsh, D.A. Mechanisms and targets of angiogenesis and nerve growth in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 390–398. [Google Scholar] [CrossRef]
- Doi, H.; Nishida, K.; Yorimitsu, M.; Komiyama, T.; Kadota, Y.; Tetsunaga, T.; Yoshida, A.; Kubota, S.; Takigawa, M.; Ozaki, T. Interleukin-4 downregulates the cyclic tensile stress-induced matrix metalloproteinases-13 and cathepsin b expression by rat normal chondrocytes. Acta Med. Okayama 2008, 62, 119–126. [Google Scholar]
- Borzi, R.M.; Olivotto, E.; Pagani, S.; Vitellozzi, R.; Neri, S.; Battistelli, M.; Falcieri, E.; Facchini, A.; Flamigni, F.; Penzo, M.; et al. Matrix metalloproteinase 13 loss associated with impaired extracellular matrix remodeling disrupts chondrocyte differentiation by concerted effects on multiple regulatory factors. Arthritis Rheumatol. 2010, 62, 2370–2381. [Google Scholar] [CrossRef]
- Houck, K.A.; Leung, D.W.; Rowland, A.M.; Winer, J.; Ferrara, N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J. Biol. Chem. 1992, 267, 26031–26037. [Google Scholar]
- Petersen, W.; Varoga, D.; Zantop, T.; Hassenpflug, J.; Mentlein, R.; Pufe, T. Cyclic strain influences the expression of the vascular endothelial growth factor (VEGF) and the hypoxia inducible factor 1 alpha (HIF-1α) in tendon fibroblasts. J. Orthop. Res. 2004, 22, 847–853. [Google Scholar] [CrossRef]
- Faure, C.; Linossier, M.T.; Malaval, L.; Lafage-Proust, M.H.; Peyroche, S.; Vico, L.; Guignandon, A. Mechanical signals modulated vascular endothelial growth factor-α (VEGF-α) alternative splicing in osteoblastic cells through actin polymerisation. Bone 2008, 42, 1092–1101. [Google Scholar] [CrossRef]
- Guignandon, A.; Usson, Y.; Laroche, N.; Lafage-Proust, M.H.; Sabido, O.; Alexandre, C.; Vico, L. Effects of intermittent or continuous gravitational stresses on cell-matrix adhesion: Quantitative analysis of focal contacts in osteoblastic ros 17/2.8 cells. Exp. Cell Res. 1997, 236, 66–75. [Google Scholar] [CrossRef]
- Sato, K.; Adachi, T.; Matsuo, M.; Tomita, Y. Quantitative evaluation of threshold fiber strain that induces reorganization of cytoskeletal actin fiber structure in osteoblastic cells. J. Biomech. 2005, 38, 1895–1901. [Google Scholar] [CrossRef]
- Liang, J.; Han, F.; Chen, Y. Transcriptional regulation of VEGF expression by estrogen-related receptor γ. Acta Pharm. Sin. B 2013, 3, 373–380. [Google Scholar] [CrossRef]
- Leychenko, A.; Konorev, E.; Jijiwa, M.; Matter, M.L. Stretch-induced hypertrophy activates NFκB-mediated VEGF secretion in adult cardiomyocytes. PLoS One 2011, 6, e29055. [Google Scholar]
- Wilkins, J.R.; Pike, D.B.; Gibson, C.C.; Kubota, A.; Shiu, Y.-T. Differential effects of cyclic stretch on BFGF- and VEGF-induced sprouting angiogenesis. Biotechnol. Prog. 2014, 30, 879–888. [Google Scholar] [CrossRef]
- Naim, R.; Sadick, H.; Bayerl, C.; Riedel, F.; Schafer, C.; Bran, G.; Hormann, K. Hepatocyte growth factor/scatter factor induces VEGF in human external auditory canal cholesteatoma cell culture. Int. J. Mol. Med. 2005, 15, 67–71. [Google Scholar]
- Haq, F.; Keith, C.; Zhang, G. Neurite development in pc12 cells on flexible micro-textured substrates under cyclic stretch. Biotechnol. Prog. 2006, 22, 133–140. [Google Scholar] [CrossRef]
- Pages, G.; Pouyssegur, J. Transcriptional regulation of the vascular endothelial growth factor gene—A concert of activating factors. Cardiovasc. Res. 2005, 65, 564–573. [Google Scholar] [CrossRef]
- Shyu, K.G.; Chang, M.L.; Wang, B.W.; Kuan, P.; Chang, H. Cyclical mechanical stretching increases the expression of vascular endothelial growth factor in rat vascular smooth muscle cells. J. Formos. Med. Assoc. 2001, 100, 741–747. [Google Scholar]
- Sun, D.; Liu, W.; Guo, K. The proximal promoter region of the human vascular endothelial growth factor gene has a G-quadruplex structure that can be targeted by G-quadruplex-interactive agents. Mol. Cancer Ther. 2008, 7, 880–888. [Google Scholar]
- Blain, E.J. Mechanical regulation of matrix metalloproteinases. Front. Biosci. 2007, 12, 507–527. [Google Scholar] [CrossRef]
- Kim, C.H.; Cho, Y.S.; Chun, Y.S.; Park, J.W.; Kim, M.S. Early expression of myocardial HIF-1α in response to mechanical stresses: Regulation by stretch-activated channels and the phosphatidylinositol 3-kinase signaling pathway. Circ. Res. 2002, 90, E25–E33. [Google Scholar] [CrossRef]
- Thomas, R.S.; Clarke, A.R.; Duance, V.C.; Blain, E.J. Effects of WNT3A and mechanical load on cartilage chondrocyte homeostasis. Arthritis Res. Ther. 2011, 13, R203. [Google Scholar] [CrossRef]
- Grad, S.; Eglin, D.; Alini, M.; Stoddart, M.J. Physical stimulation of chondrogenic cells in vitro: A review. Clin. Orthop. Relat. Res. 2011, 469, 2764–2772. [Google Scholar] [CrossRef]
- Kasuno, K.; Takabuchi, S.; Fukuda, K.; Kizaka-Kondoh, S.; Yodoi, J.; Adachi, T.; Semenza, G.L.; Hirota, K. Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on mapk and phosphatidylinositol 3-kinase signaling. J. Biol. Chem. 2004, 279, 2550–2558. [Google Scholar]
- Olsson, A.K.; Dimberg, A.; Kreuger, J.; Claesson-Welsh, L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006, 7, 359–371. [Google Scholar] [CrossRef]
- Jansen, J.H.; Weyts, F.A.; Westbroek, I.; Jahr, H.; Chiba, H.; Pols, H.A.; Verhaar, J.A.; van Leeuwen, J.P.; Weinans, H. Stretch-induced phosphorylation of ERK1/2 depends on differentiation stage of osteoblasts. J. Cell. Biochem. 2004, 93, 542–551. [Google Scholar] [CrossRef]
- Jansen, J.H.; Jahr, H.; Verhaar, J.A.; Pols, H.A.; Chiba, H.; Weinans, H.; van Leeuwen, J.P. Stretch-induced modulation of matrix metalloproteinases in mineralizing osteoblasts via extracellular signal-regulated kinase-1/2. J. Orthop. Res. 2006, 24, 1480–1488. [Google Scholar] [CrossRef]
- Mizuno, S. A novel method for assessing effects of hydrostatic fluid pressure on intracellular calcium: A study with bovine articular chondrocytes. Am. J. Physiol. Cell. Physiol. 2005, 288, C329–C337. [Google Scholar] [CrossRef]
- Farhang Ghahremani, M.; Goossens, S.; Haigh, J.J. The p53 family and vegf regulation: “It’s complicated”. Cell Cycle 2013, 12, 1331–1332. [Google Scholar]
- Knight, M.M.; van de Breevaart Bravenboer, J.; Lee, D.A.; van Osch, G.J.; Weinans, H.; Bader, D.L. Cell and nucleus deformation in compressed chondrocyte-alginate constructs: Temporal changes and calculation of cell modulus. Biochim. Biophys. Acta 2002, 1570, 1–8. [Google Scholar]
- Greiner, A.M.; Chen, H.; Spatz, J.P.; Kemkemer, R. Cyclic tensile strain controls cell shape and directs actin stress fiber formation and focal adhesion alignment in spreading cells. PLoS One 2013, 8, e77328. [Google Scholar]
- Yu, J.; Xie, Y.J.; Xu, D.; Zhao, S.L. Effect of cyclic strain on cell morphology, viability and proliferation of human dental pulp cells in vitro. Shanghai Kou Qiang Yi Xue 2009, 18, 599–603. [Google Scholar]
- Aigner, T.; Haag, J.; Martin, J.; Buckwalter, J. Osteoarthritis: Aging of matrix and cells—Going for a remedy. Curr. Drug Targets 2007, 8, 325–331. [Google Scholar] [CrossRef]
- Goldring, M.B.; Birkhead, J.R.; Suen, L.F.; Yamin, R.; Mizuno, S.; Glowacki, J.; Arbiser, J.L.; Apperley, J.F. Interleukin-1β-modulated gene expression in immortalized human chondrocytes. J. Clin. Investig. 1994, 94, 2307–2316. [Google Scholar]
- Finger, F.; Schorle, C.; Zien, A.; Gebhard, P.; Goldring, M.B.; Aigner, T. Molecular phenotyping of human chondrocyte cell lines t/c-28a2, t/c-28a4, and c-28/i2. Arthritis Rheumatol. 2003, 48, 3395–3403. [Google Scholar] [CrossRef]
- Fragoulis, A.; Laufs, J.; Muller, S.; Soppa, U.; Siegl, S.; Reiss, L.K.; Tohidnezhad, M.; Rosen, C.; Tenbrock, K.; Varoga, D.; et al. Sulforaphane has opposing effects on TNF-α stimulated and unstimulated synoviocytes. Arthritis Res. Ther. 2012, 14, R220. [Google Scholar]
- Van de Werken, C.; Jahr, H.; Avo Santos, M.; Eleveld, C.; Schuilwerve, J.; Laven, J.S.; Baart, E.B. A universal method for sequential immunofluorescent analysis of chromatin and chromatin-associated proteins on chromosome spreads. Chromosom. Res. 2013, 21, 475–489. [Google Scholar]
- Burgess, A.; Vigneron, S.; Brioudes, E.; Labbe, J.C.; Lorca, T.; Castro, A. Loss of human greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin b-cdc2/pp2a balance. Proc. Natl. Acad. Sci. USA 2010, 107, 12564–12569. [Google Scholar]
- Science Tech Blog. Available online: http://sciencetechblog.com/2011/05/24/measuring-cell-fluorescence-using-imagej/ (accessed on 28 August 2014).
- ImageJ. Available online: http://imagej.nih.gov/ij/ (accessed on 28 August 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Beckmann, R.; Houben, A.; Tohidnezhad, M.; Kweider, N.; Fragoulis, A.; Wruck, C.J.; Brandenburg, L.O.; Hermanns-Sachweh, B.; Goldring, M.B.; Pufe, T.; et al. Mechanical Forces Induce Changes in VEGF and VEGFR-1/sFlt-1 Expression in Human Chondrocytes. Int. J. Mol. Sci. 2014, 15, 15456-15474. https://doi.org/10.3390/ijms150915456
Beckmann R, Houben A, Tohidnezhad M, Kweider N, Fragoulis A, Wruck CJ, Brandenburg LO, Hermanns-Sachweh B, Goldring MB, Pufe T, et al. Mechanical Forces Induce Changes in VEGF and VEGFR-1/sFlt-1 Expression in Human Chondrocytes. International Journal of Molecular Sciences. 2014; 15(9):15456-15474. https://doi.org/10.3390/ijms150915456
Chicago/Turabian StyleBeckmann, Rainer, Astrid Houben, Mersedeh Tohidnezhad, Nisreen Kweider, Athanassios Fragoulis, Christoph J. Wruck, Lars O. Brandenburg, Benita Hermanns-Sachweh, Mary B. Goldring, Thomas Pufe, and et al. 2014. "Mechanical Forces Induce Changes in VEGF and VEGFR-1/sFlt-1 Expression in Human Chondrocytes" International Journal of Molecular Sciences 15, no. 9: 15456-15474. https://doi.org/10.3390/ijms150915456