Expression of Aspergillus nidulans phy Gene in Nicotiana benthamiana Produces Active Phytase with Broad Specificities

Abstract
:1. Introduction
2. Results and Discussion
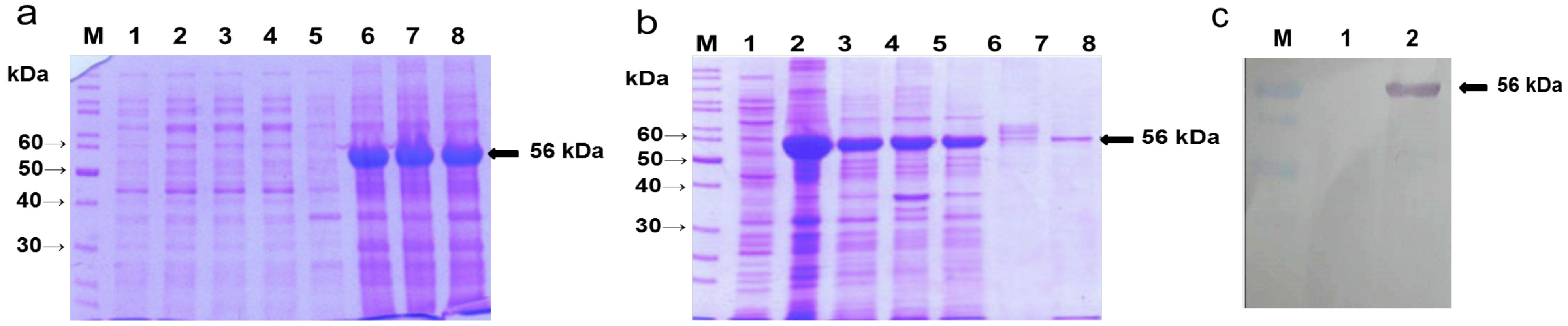
2.1. Overexpression of A. nidulans phy Gene in E. coli, Purification of the Phy-6x His-Tagged Fusion Protein and Production of the Recombinant Protein (rPhy-E)-Specific Antibody


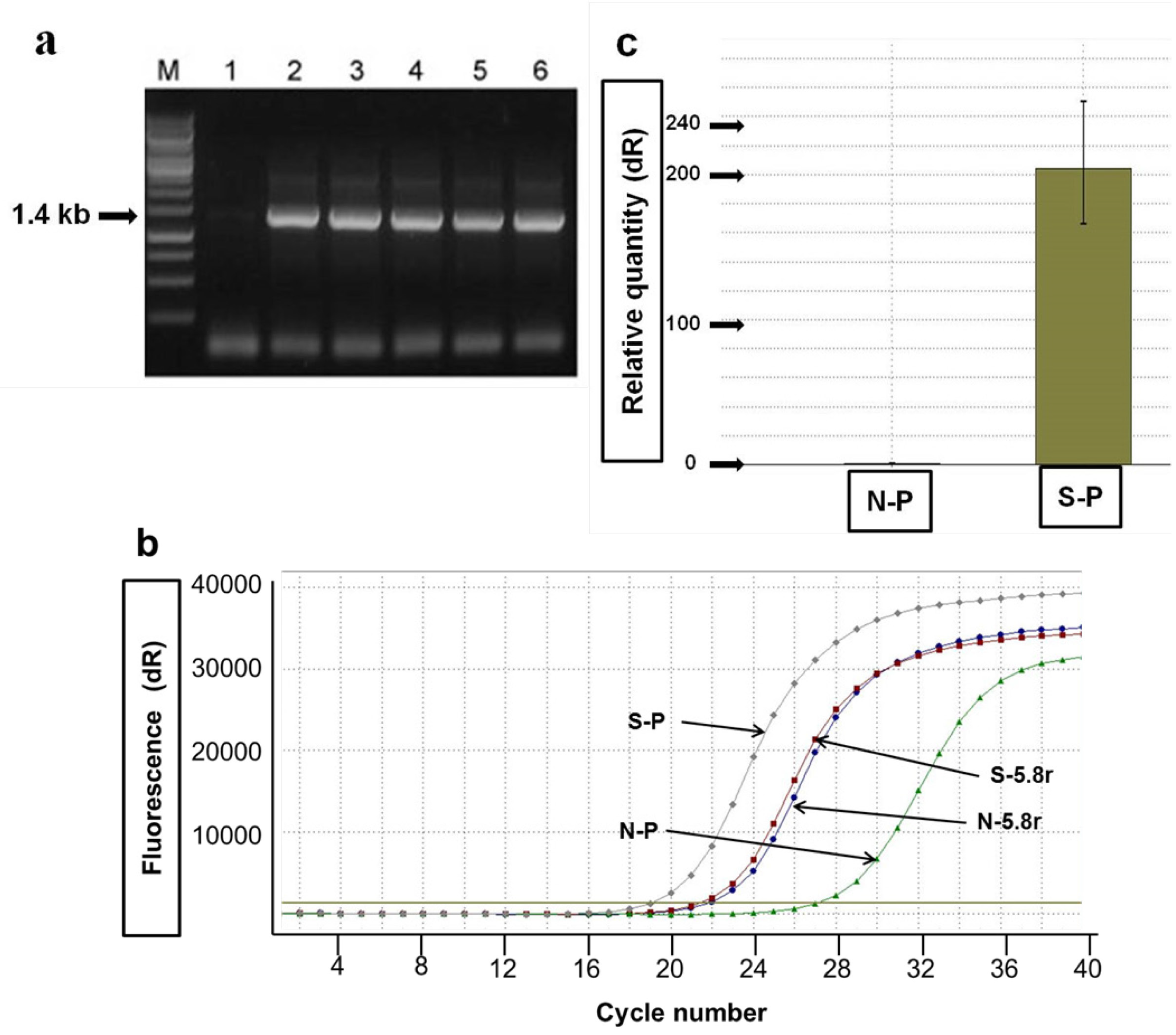
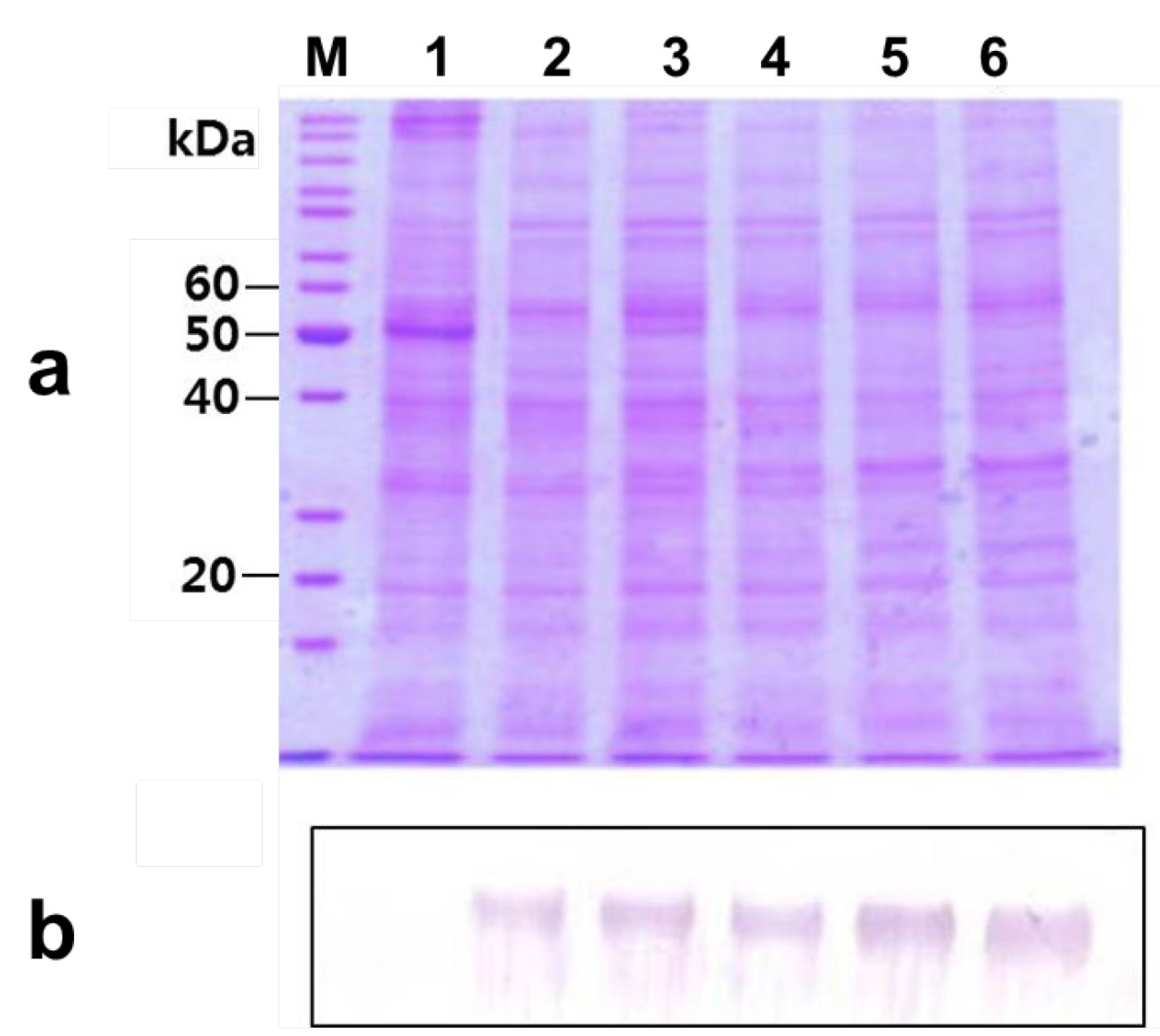
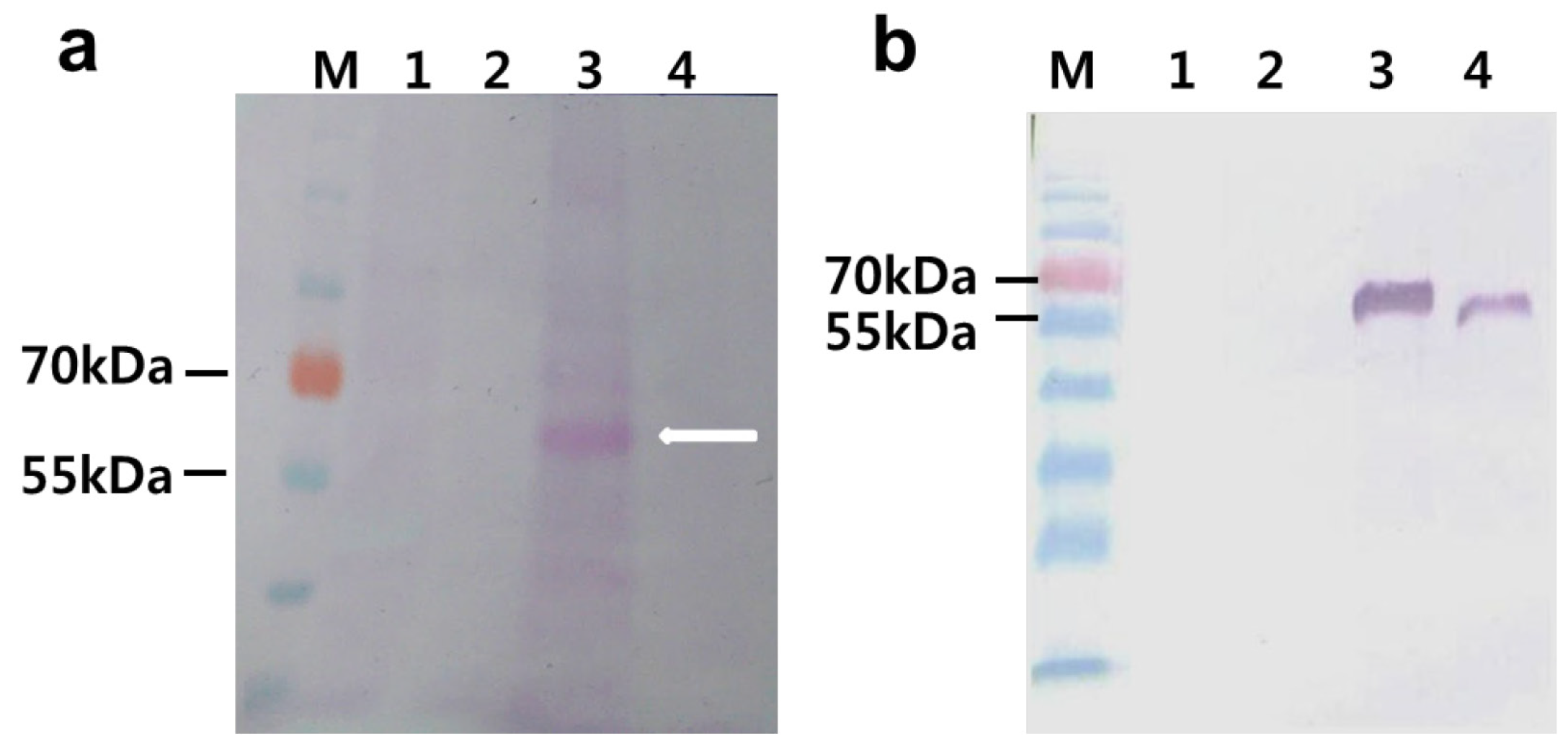
2.2. Agro-Infiltration and phy Gene Expression in N. benthamiana

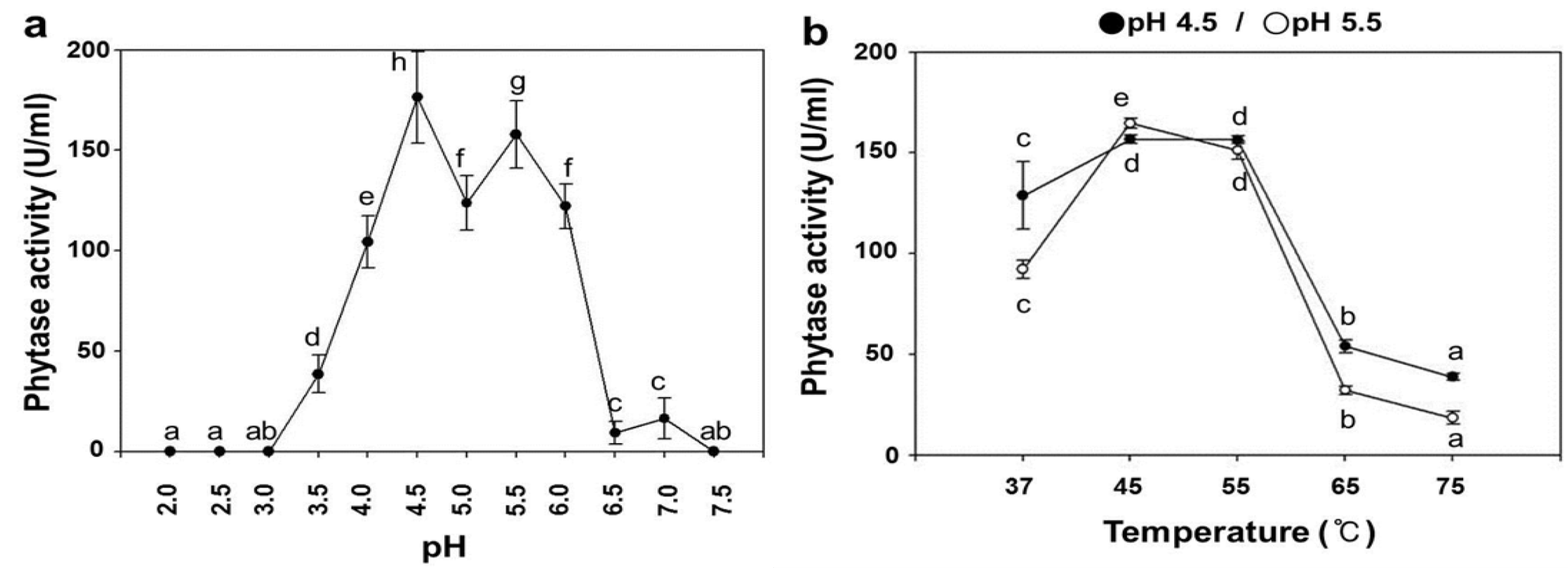
2.3. pH Dependence on the Phytase Activity of rPhy-P

2.4. Temperature Dependence on the Phytase Activity of rPhy-P

2.5. Deglycosylation of the Expressed Phytase
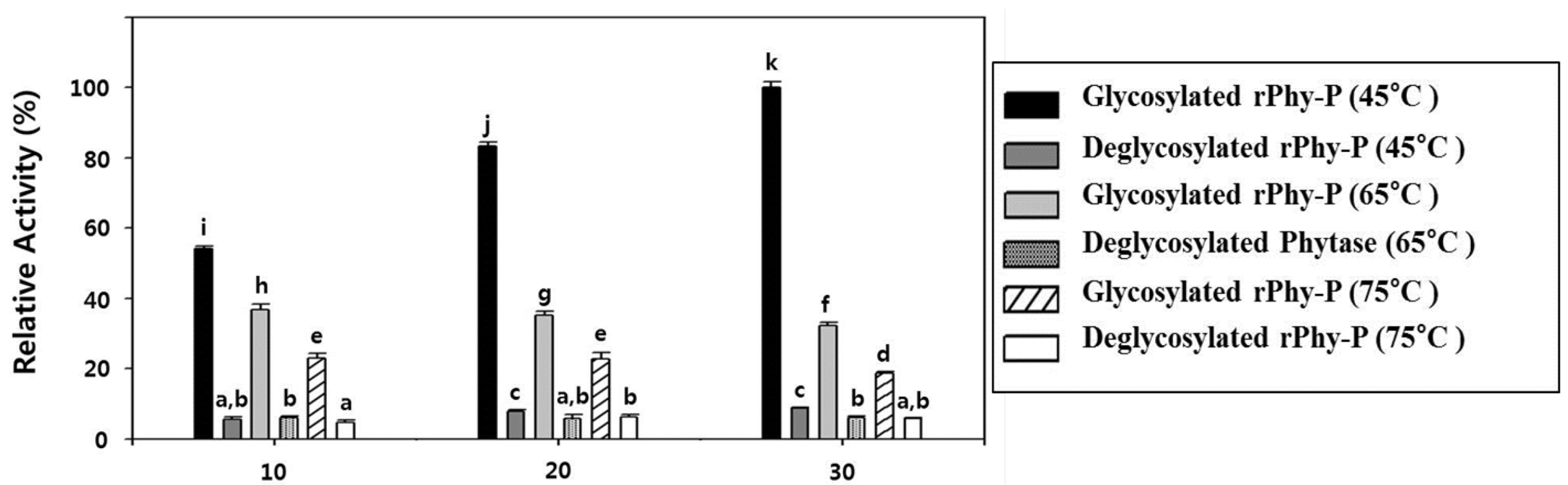
2.6. Glycosylation Effect on the Thermostability of the Phytase Activity of rPhy-P

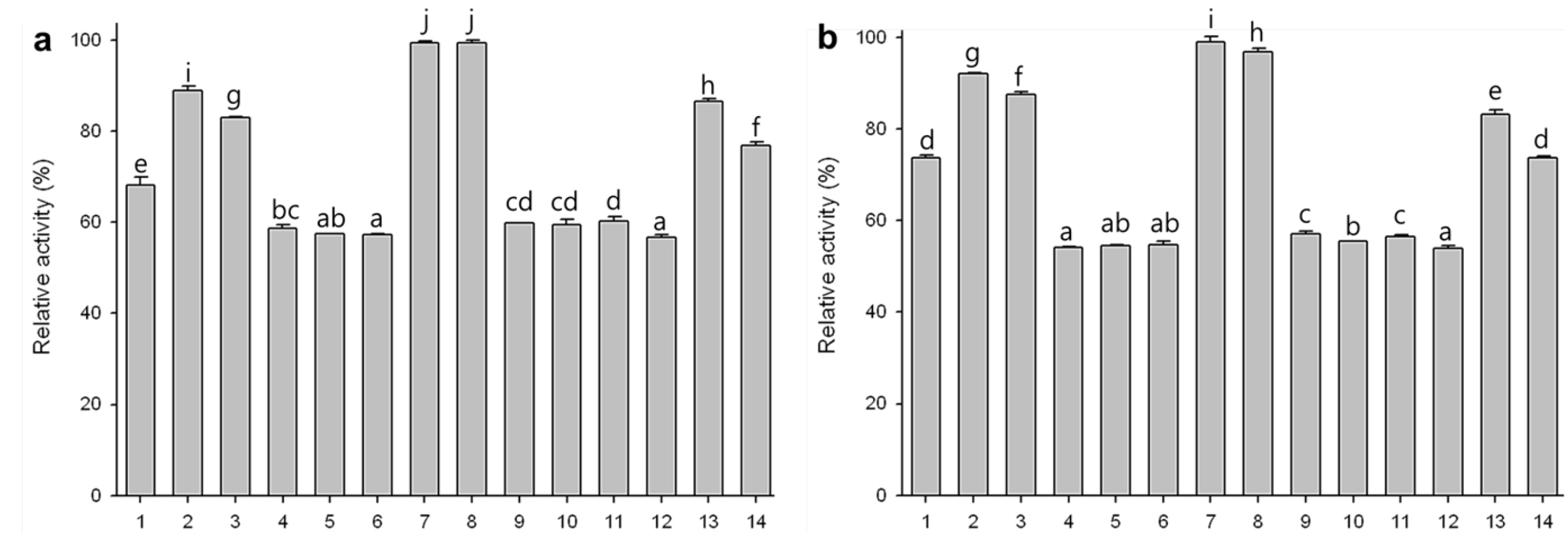
2.7. Substrate Specificities of Phytase Expressed in Agro-Infiltrated Leaves


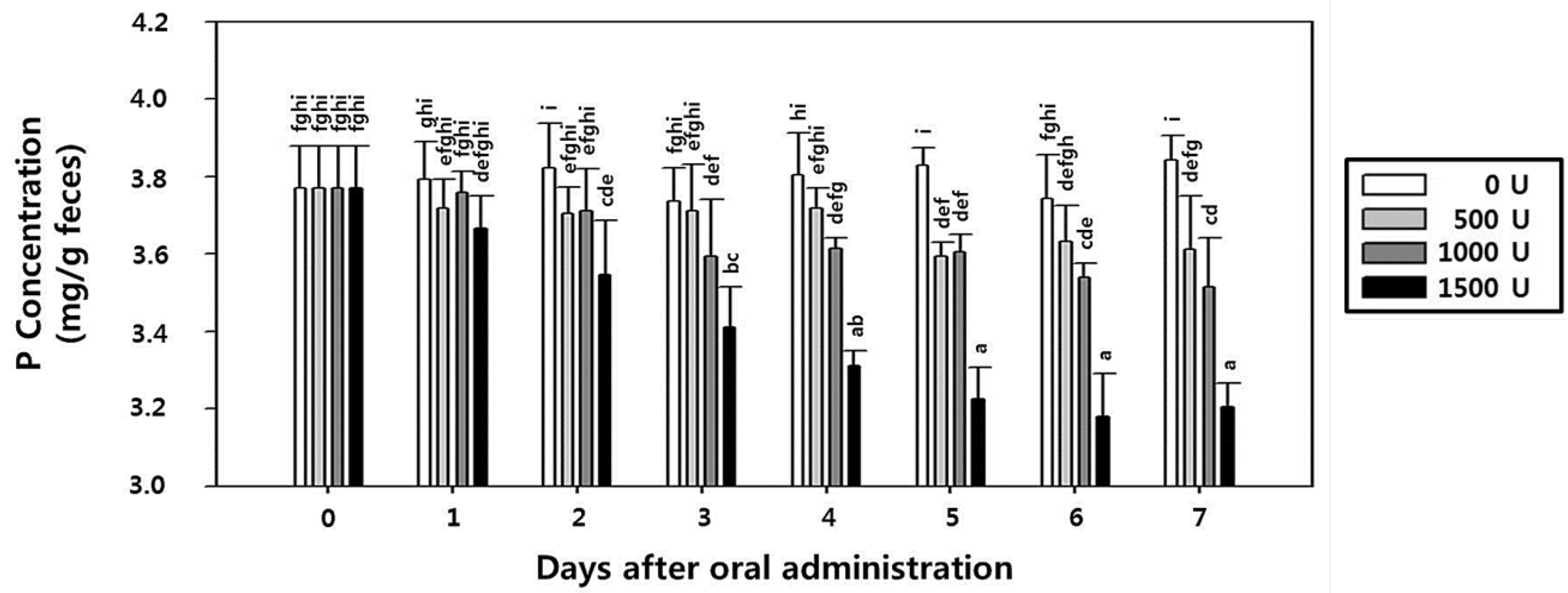
2.8. Effect of rPhy-P on Phosphorus Reduction and Toxicity in Rat Fecal Samples
3. Experimental Section
3.1. Computer-Aided Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Template | Primer | Nucleotide Sequences (5'→3') |
|---|---|---|
| cDNA libraries | phy1-F 1 | GAATTCGCTTTTTTCACGGTCGCTCTT |
| phy1-R 2 | GTCGACTAGGGTAAAACAAGTCTTCCA | |
| pRTL2 | M1-RTL-F 3 | GAAAATTTTCACCATTTACGAACGATAGCCGCGGCACTCATC |
| M1-RTL-R 4 | GATGAGTGCCGCGGCTATCGTTCGTAAATGGTGAAAATTTTC | |
| pET-phy | phy2-F 5 | CCGCGGCAATGGCTTTTTTCACG |
| phy2-R 6 | GGTACCTTATAGGGTAAAACAAGTCTTCCA | |
| pRTL2-MS-Phytase | M2-RTL-F 7 | TTACGAACGATAGCCATGGCAATGGCTTTTTTC |
| M2-RTL-R 8 | GAAAAAAGCCATTGCCATGGCTATCGTTCGTAA | |
| pPZP212-phy | phy3-F | CCGCGGCATCTCATGTTTGGGGT |
| phy3-R | TCTAGAGGGGCTTCCGGCACC | |
| N. benthamiana total RNA (mock and agroinfitrated) | phy-domain-F 9 | CAGAGTCGAAGAGTAAGGCGTAC |
| phy-domain-R 9 | GTAGAACTTGGCACCCGAATCAAC | |
| N. benthamiana total RNA (mock and agroinfitrated) | 5.8s rRNA-F 10 | GTGATCTGTGGAAGGATCATTGTCG |
| 5.8s rRNA-R 10 | CGTTAATCATCCGACACGAACGC |
3.2. PCR and Cloning of the phy Gene of A. nidulans into pET28a
3.3. Expression and Purification of His-Tagged Recombinant Protein pET28a
3.4. Production of rPhy-E Specific Polyclonal Antibody
3.5. PCR Amplification of phy Gene and Vector Construction for Plant Expression
3.6. Agrobacterium Transformation, Agro-Infiltration and Analysis by RT-PCR and Quantitative Real-Time RT-PCR
3.7. SDS-PAGE, Western Blot and Deglycosylation Analysis of Protein Extracts
3.8. Phytase Assay and Substrate Specificities
3.9. Determination of Optimal pH and Temperature for Phytase Activity
3.10. Determination of Thermostability before and after Deglycosylation
3.11. Experimental Animal Trial and Design
3.12. Determination of Phosphorus (P) in Fecal Samples
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lei, X.G.; Porres, J.M.; Mullaney, E.J.; Brinch-Pedersen, H. Phytase: Source, structure and application. In Industrial Enzymes Structure, Function and Applications; MacCabe, A.P., Polaina, J., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 505–529. [Google Scholar]
- Singh, B.; Gotthard Kunze, G.; Satyanarayana, T. Developments in biochemical aspects and biotechnological applications of microbial phytases. Biotechnol. Mol. Biol. Rev. 2011, 6, 69–87. [Google Scholar]
- Al-Asheh, S.; Duvnjak, Z. Characteristics of phytase produced by Aspergillus carbonarius NRC 401121 in canola meal. Acta Biotechnol. 1994, 14, 223–233. [Google Scholar] [CrossRef]
- Casey, A.; Walsh, G. Purification and characterization of extracellular phytase from Aspergillus niger ATCC 9142. Bioresour. Technol. 2003, 86, 183–188. [Google Scholar] [CrossRef]
- Fujita, J.; Yamane, Y.-I.; Fukuda, H.; Kizaki, Y.; Wakabayashi, S.; Shigeta, S.; Suzuki, O.; Ono, K. Production and properties of phytase and acid phosphotase from a sake koji mold, Aspergillus oryzae. J. Biosci. Bioeng. 2003, 95, 348–353. [Google Scholar]
- Pasamontes, L.; Haiker, M.; Wyss, M.; Tessier, M.; van Loon, A.P. Gene cloning, purification, and characterization of a heat-stable phytase from the fungus Aspergillus fumigatus. Appl. Environ. Microbiol. 1997, 63, 1696–1700. [Google Scholar]
- Ullah, A.H.J.; Cummins, B.J. Purification, N-terminal amino acid sequence and characterization of the pH 2.5 optimum acid phosphatase (E.C. 3.1.3.2) from Aspergillus ficuum. Prep. Biochem. 1987, 17, 397–422. [Google Scholar]
- Ullah, A.H.J.; Cummins, B.J. Aspergillus ficuum extracellular pH 6.0 optimum acid phosphatase: Purification, N-terminal amino acid sequence, and biochemical characterization. Prep. Biochem. 1988, 18, 37–65. [Google Scholar]
- Ullah, A.H.J.; Gibson, D.M. Extracellular phytase (E.C. 3.1.3.8) from Aspergillus ficuum NRRL 3135: purification and characterization. Prep. Biochem. 1987, 17, 63–91. [Google Scholar]
- Vats, P.; Banerjee, U.C. Biochemical characterisation of extracellular phytase (myo-inositol hexakisphosphate phosphohydrolase) from a hyper-producing strain of Aspergillus niger van Teighem. J. Ind. Microbiol. Biotechnol. 2005, 32, 141–147. [Google Scholar]
- Zhang, G.Q.; Dong, X.F.; Wang, Z.H.; Zhang, Q.; Wang, H.X.; Tong, J.M. Purification, characterization, and cloning of a novel phytase with low pH optimum and strong proteolysis resistance from Aspergillus ficuum NTG-23. Bioresour. Technol. 2010, 101, 4125–4131. [Google Scholar]
- Lei, X.G.; Porres, J.M. Phytase enzymology, applications, and biotechnology. Biotechnol. Lett. 2003, 25, 1787–1794. [Google Scholar] [CrossRef]
- Wodzinski, R.J.; Ullah, A.H.J. Phytase. Adv. Appl. Microbiol. 1996, 42, 263–302. [Google Scholar] [CrossRef]
- Wyss, M.; Pasamontes, L.; Remy, R.; Kohler, J.; Kusznir, E.; Gadient, M.; Muller, F.; van Loon, A.P.G.M. Comparison of the thermostability properties of three acid phosphatases from molds: Aspergillus fumigatus phytase, A. niger phytase, and A. niger pH 2.5 acid phosphatase. Appl. Environ. Microbiol. 1998, 64, 4446–4451. [Google Scholar]
- Wyss, M.; Brugger, R.; Kronenberger, A.; Remy, R.; Fimbel, R.; Oesterhelt, G.; Lehmann, M.; van Loon, A.P.G.M. Biochemical characterization of fungal phytases (myo-inositol hexakisphosphate phosphohydrolases): Catalytic properties. Appl. Environ. Microbiol. 1999, 65, 367–373. [Google Scholar]
- Kostrewa, D.; Wyss, M.; D’Arcy, A.; van Loon, A.P.G.M. Crystal structure of Aspergillus niger pH 2.5 acid phosphatase at 2.4 Å resolution. J. Mol. Biol. 1999, 288, 965–974. [Google Scholar]
- Mullaney, E.J.; Ullah, A.H.J. The term phytase comprises several different classes of enzymes. Biochem. Biophys. Res. Commun. 2003, 312, 179–184. [Google Scholar]
- Han, Y.; Lei, X.G. Role of glycosylation in the functional expression of an Aspergillus niger phytase gene (phyA) in Pichia pastoris. Arch. Biochem. Biophys. 1999, 364, 83–90. [Google Scholar]
- Han, Y.; Wilson, D.B.; Lei, X. Expression of an Aspergillus niger phytase gene (phyA) in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 1999, 65, 1915–1918. [Google Scholar]
- Rodriguez, E.; Mullaney, E.J.; Lei, X.G. Expression of Aspergillus fumigatus phytase gene in Pichia pastoris and characterization of the recombinant enzyme. Biochem. Biophys. Res. Commun. 2000, 268, 373–378. [Google Scholar]
- Guo, M.; Hang, H.; Zhu, T.; Zhuang, Y.; Chu, J.; Zhang, S. Effect of glycosylation on biochemical characterization of recombinant phytase expressed in Pichia pastoris. Enzym. Microb. Technol. 2008, 42, 340–345. [Google Scholar]
- Liu, J.F.; Wang, X.F.; Li, Q.L.; Li, X.; Zhang, G.Y.; Li, M.G.; Ma, Z.Y. Biolistic transformation of cotton (Gossypium hirsutum L.) with the phyA gene from Aspergillus ficuum. Plant Cell Tissue Organ Cult. 2011, 106, 207–214. [Google Scholar] [CrossRef]
- Richardson, A.E.; Hadobas, P.A.; Hayes, J.E. Extracellular secretion of Aspergillus phytase from Arabidopsis roots enables plants to obtain phosphorus from phytate. Plant J. 2001, 25, 641–649. [Google Scholar]
- Brinch-Pedersen, H.; Hatzack, F.; Sørensen, L.D.; Holm, P.B. Concerted action of endogenous and heterologous phytase on phytic acid degradation in seed of transgenic wheat (Triticum aestivum L.). Transgenic Res. 2003, 12, 649–659. [Google Scholar]
- Chen, R.; Xue, G.; Chen, P.; Yao, B.; Yang, W.; Ma, Q.; Fan, Y.; Zhao, Z.; Tarczynski, M.C.; Shi, J. Transgenic maize plants expressing a fungal phytase gene. Transgenic Res. 2008, 17, 633–643. [Google Scholar]
- Drakakaki, G.; Marcel, S.; Glahn, R.P.; Lund, E.K.; Pariagh, S.; Fisher, R.; Christou, P.; Stoger, E. Endosperm-specific co-expression of recombinant soybean ferritin and Aspergillus phytase in maize results in significant increases in the levels of bioavailable iron. Plant Mol. Biol. 2005, 59, 869–880. [Google Scholar]
- George, T.S.; Richardson, A.E.; Hadobas, P.A.; Simpson, R.J. Characterisation of transgenic Trifolium subterraneum L. which expresses phyA and releases extracellular phytase: growth and P nutrition in laboratory media and soil. Plant Cell Environ. 2004, 27, 1351–1361. [Google Scholar] [CrossRef]
- George, T.S.; Simpson, R.J.; Hadobas, P.A.; Richardson, A.E. Expression of a fungal phytase gene in Nicotiana tabacum improves phosphorus nutrition of plants grown in amended soils. Plant Biotechnol. J. 2005, 3, 129–140. [Google Scholar]
- Lucca, P.; Hurrell, R.; Potrykus, I. Genetic engineering approaches to improve the bioavailability and the level of iron in rice grains. Theor. Appl. Genet. 2001, 102, 392–397. [Google Scholar]
- Ponstein, A.S.; Bade, J.B.; Verwoerd, T.C.; Molendijk, L.; Storms, J.; Beudeker, R.F.; Pen, J. Stable expression of phytase (phyA) in canola (Brassica napus) seeds: towards a commercial product. Mol. Breed. 2002, 10, 31–44. [Google Scholar] [CrossRef]
- Ullah, A.H.J.; Sethumadhavan, K.; Mullaney, E.J.; Ziegelhoffer, T.; Austin-Phillips, S. Cloned and expressed fungal phyA gene in alfalfa produces a stable phytase. Biochem. Biophys. Res. Commun. 2002, 290, 1343–1348. [Google Scholar]
- Ullah, A.H.J.; Sethumadhavan, K.; Mullaney, E.J.; Ziegelhoffer, T.; Austin-Phillips, S. Fungal phyA gene expressed in potato leaves produces active and stable phytase. Biochem. Biophys. Res. Commun. 2003, 306, 603–609. [Google Scholar]
- Xiao, K.; Katagi, H.; Harrison, M.; Wang, Z.-Y. Improved phosphorus acquisition and biomass production in Arabidopsis by transgenic expression of a purple acid phosphatase gene from M. truncatula. Plant Sci. 2006, 170, 191–202. [Google Scholar]
- Pasamontes, L.; Haiker, M.; Henriquez-Huecas, M.; Mitchell, D.B.; van Loon, A.P.G.M. Cloning of the phytases from Emericella nidulans and the thermophilic fungus Talaromyces thermophilus. Biochim. Biophys. Acta 1997, 1353, 217–223. [Google Scholar]
- Wyss, M.; Pasamontes, L.; Friedlein, A.; Remy, R.; Tessier, M.; Kronenberger, A.; Middendorf, A.; Lehmann, M.; Schnoebelen, L.; Röthlisberger, U.; et al. Biophysical characterization of fungal phytases (myo-inositol hexakisphosphate phosphohydrolases): Molecular size, glycosylation pattern, and engineering of proteolytic resistance. Appl. Environ. Microbiol. 1999, 65, 359–366. [Google Scholar]
- Kniskern, P.J.; Hagopian, A.; Burke, P.; Schultz, L.D.; Montgomery, D.L.; Hurni, W.M.; Ip, C.Y.; Schulman, C.A.; Maigetter, R.Z.; Wampler, D.E.; et al. Characterization and evaluation of a recombinant hepatitis B vaccine expressed in yeast defective for N-linked hyperglycosylation. Vaccine 1994, 12, 1021–1025. [Google Scholar] [CrossRef]
- Phillippy, B.Q.; Mullaney, E.J. Expression of an Aspergillus niger phytase (phyA) in Escherichia coli. J. Agric. Food Chem. 1997, 45, 3337–3342. [Google Scholar]
- Kim, S.-Y.; Sohn, J.-H.; Pyun, Y.-R.; Choi, E.-S. Variations in protein glycosylation in Hansenula polymorpha depending on cell culture stage. J. Microbiol. Biotechnol. 2007, 17, 1949–1954. [Google Scholar]
- Lee, J.; Park, J.-S.; Moon, J.-Y.; Kim, K.-Y; Moon, H.-M. The influence of glycosylation on secretion, stability, and immunogenicity of recombinant HBV pre-S antigen synthesized in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2003, 303, 427–432. [Google Scholar]
- Lam, J.S.; Huang, H.; Levitz, S.M. Effect of differential N-linked and O-linked mannosylation on recognition of fungal antigens by dendritic cells. PLoS One 2007, 2, e1009. [Google Scholar]
- Dasgupta, S.; Navarrete, A.M.; Bayry, J.; Delignat, S.; Wootla, B.; Andre, S.; Christophe, O.; Nascimbeni, M.; Jacquemin, M.; Martinez-Pomares, L.; et al. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 8965–8970. [Google Scholar]
- Li, H.; D’Anjou, M. Pharmacological significance of glycosylation in therapeutic proteins. Curr. Opin. Biotechnol. 2009, 29, 678–684. [Google Scholar]
- Schmidt, F.R. Recombinant expression systems in the pharmaceutical industry. Appl. Microbiol. Biotechnol. 2004, 65, 363–372. [Google Scholar] [CrossRef]
- Demain, A.L.; Vaishnav, P. Production of recombinant proteins by microbes and higher organisms. Biotechnol. Adv. 2009, 27, 297–306. [Google Scholar] [CrossRef]
- Walsh, G. Biopharmaceutical benchmarks. Nat. Biotechnol. 2003, 21, 865–870. [Google Scholar] [CrossRef]
- Bosch, D.; Castilho, A.; Loos, A.; Schots, A.; Steinkellner, H. N-glycosylation of plant-produced recombinant proteins. Curr. Pharm. Des. 2013, 19, 5503–5512. [Google Scholar]
- Casey, A.; Walsh, G. Identification and characterization of a phytase of potential commercial interest. J. Biotechnol. 2004, 110, 313–322. [Google Scholar] [CrossRef]
- Protein blast. Blastp Program. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 26 June 2012).
- Compute pI/MW Tool. Available online: http://web.expasy.org/computre_pi (accessed on 26 June 2012).
- Cho, J.-H.; Yun, S.-S.; Jang, Y.-K.; Cha, M.-J.; Kwon, N.-J.; Chae, S.-K. Identification and cloning of jipA enoding polypeptide that interacts with a homolog of yeast Rad6, UVSJ in Aspergillus nidulans. J. Microbiol. 2003, 41, 46–51. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Pfaffl, M.W. Relative quantification. In Real-Time PCR; Dorak, T., Ed.; International University Line: La Jolla, CA, USA, 2006; pp. 63–82. [Google Scholar]
- Dubray, G.; Bezard, G. A highly sensitive periodic acid-silver stain for 1,2-diol groups of glycoproteins and polysaccharides in polyacrylamide gels. Anal. Biochem. 1982, 119, 325–329. [Google Scholar]
- Holman, W.I. A new technique for the determination of phosphorus by the molybdenum blue method. Biochem. J. 1943, 37, 256–259. [Google Scholar]
- Murphy, J.; Riley, J.P. A modified single solution method for the determination of phosphate in natural waters. Anal. Chim. Acta 1962, 27, 31–36. [Google Scholar]
- Sims, J.T. Soil test Phosphorus: Mehlich 3. In Methods of Phosphorus Analysis for Soils, Sediments, Residuals, and Waters; Pierzynski, G.M., Ed.; Southern Cooperative Series Bulletin N0. 36; SAAESD, North Carolina State University: Raleigh, NC, USA, 2000; pp. 17–19. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oh, T.-K.; Oh, S.; Kim, S.; Park, J.S.; Vinod, N.; Jang, K.M.; Kim, S.C.; Choi, C.W.; Ko, S.-M.; Jeong, D.K.; et al. Expression of Aspergillus nidulans phy Gene in Nicotiana benthamiana Produces Active Phytase with Broad Specificities. Int. J. Mol. Sci. 2014, 15, 15571-15591. https://doi.org/10.3390/ijms150915571
Oh T-K, Oh S, Kim S, Park JS, Vinod N, Jang KM, Kim SC, Choi CW, Ko S-M, Jeong DK, et al. Expression of Aspergillus nidulans phy Gene in Nicotiana benthamiana Produces Active Phytase with Broad Specificities. International Journal of Molecular Sciences. 2014; 15(9):15571-15591. https://doi.org/10.3390/ijms150915571
Chicago/Turabian StyleOh, Tae-Kyun, Sung Oh, Seongdae Kim, Jae Sung Park, Nagarajan Vinod, Kyung Min Jang, Sei Chang Kim, Chang Won Choi, Suk-Min Ko, Dong Kee Jeong, and et al. 2014. "Expression of Aspergillus nidulans phy Gene in Nicotiana benthamiana Produces Active Phytase with Broad Specificities" International Journal of Molecular Sciences 15, no. 9: 15571-15591. https://doi.org/10.3390/ijms150915571