Synthesis and Characterization of New Bivalent Agents as Melatonin- and Histamine H3-Ligands


 ,
,
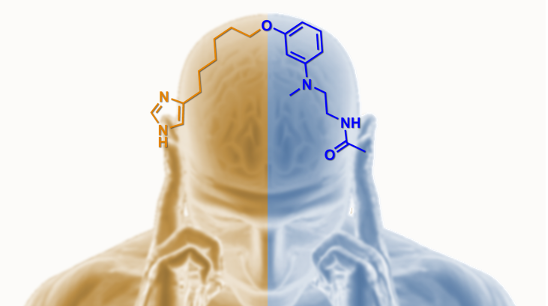
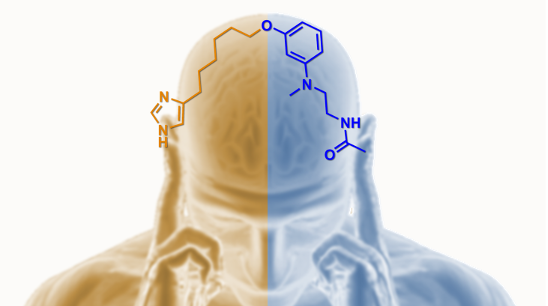
Abstract
:
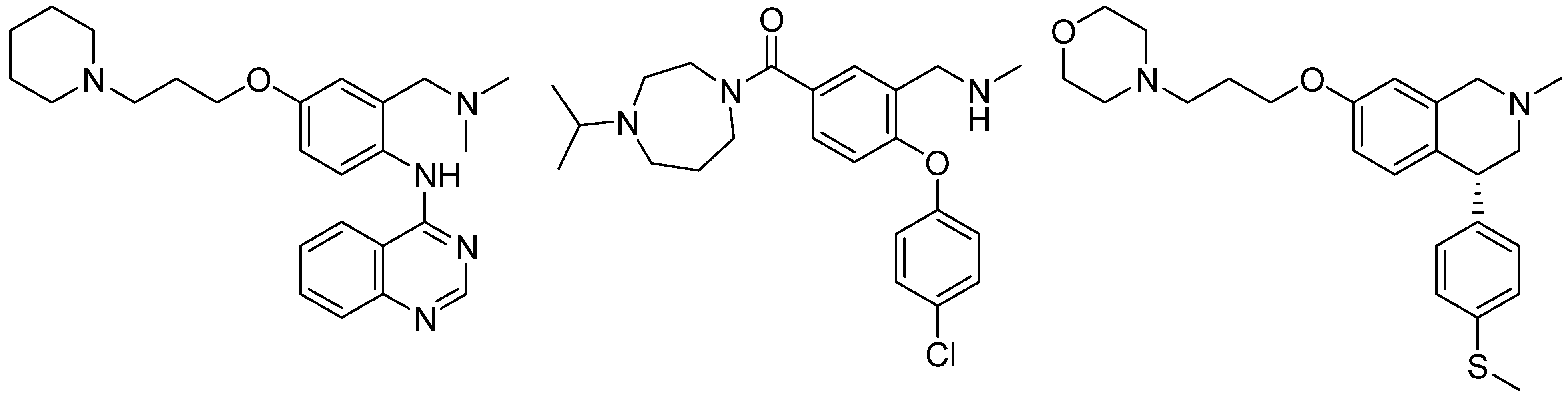
1. Introduction





2. Results and Discussion
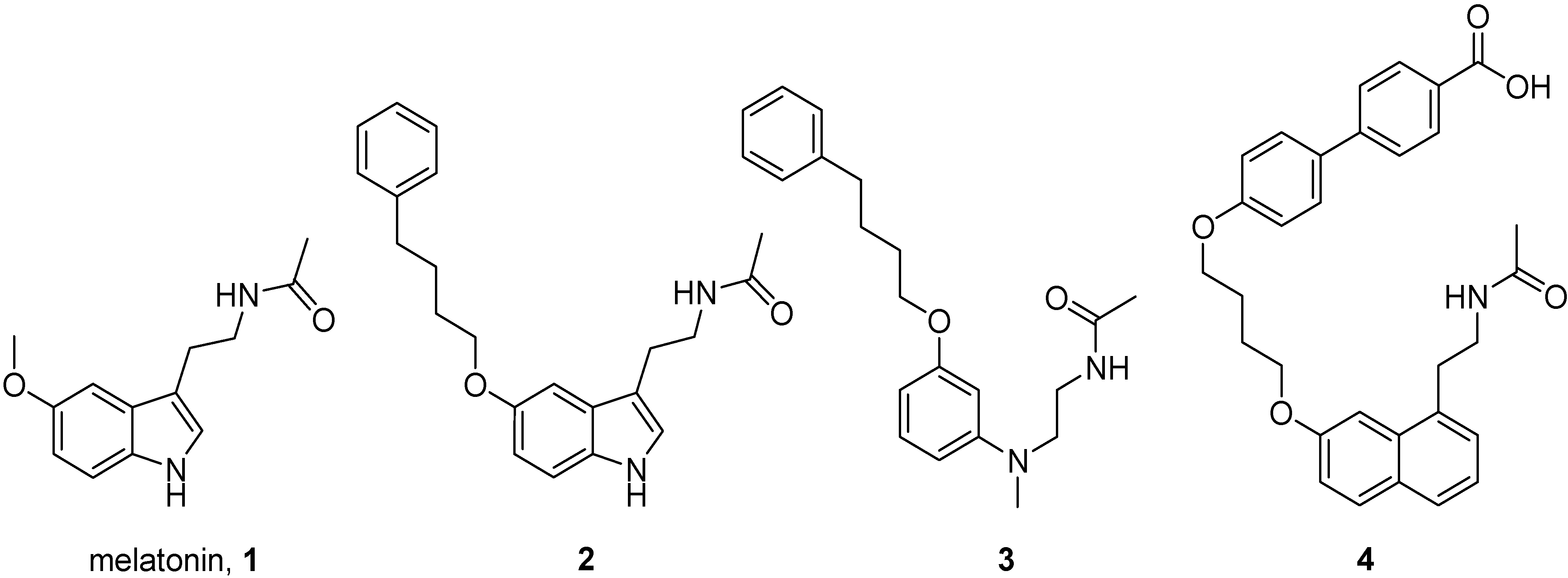
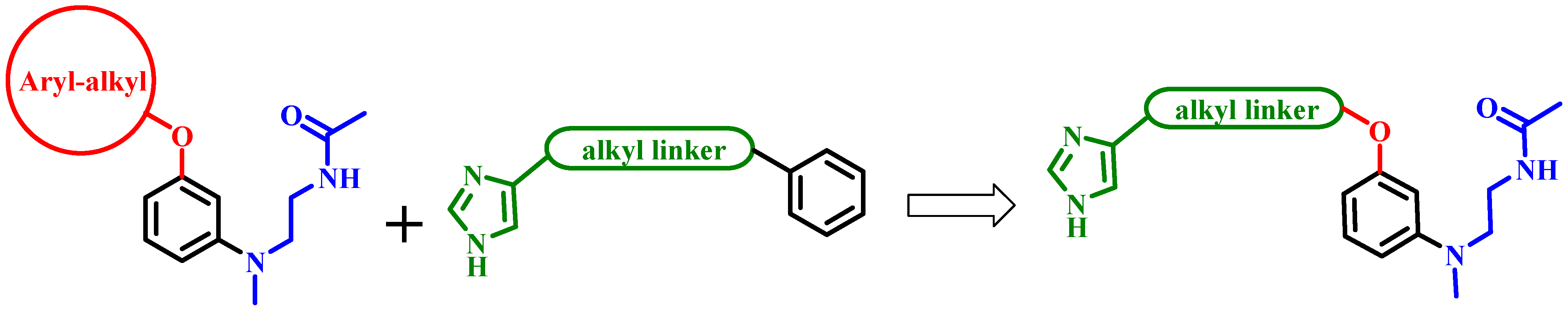
2.1. Chemistry


2.2. Binding Affinities of Compounds 14a–d for Melatonin MT1, MT2 and Histamine H3 Receptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
| Compound | n | hMT1 | hMT2 | hH3 | ||
|---|---|---|---|---|---|---|
| pKi | IAR | pKi | IAR | pKi | ||
| 1 | 9.60 ± 0.18 | 1.00 ± 0.09 | 9.44 ± 0.12 | 1.00 ± 0.07 | N.D. | |
| 5 | N.D. | N.D. | N.D. | N.D. | 7.28 ± 0.15 | |
| 14a | 0 | N.A. | N.A. | 5.91 ± 0.01 | ||
| 14b | 1 | N.A. | N.A. | N.D. | ||
| 14c | 2 | 6.09 ± 0.12 | N.D. | 6.28 ± 0.10 | N.D. | 6.28 ± 0.03 |
| 14d | 3 | 6.79 ± 0.01 | −0.26 ± 0.09 | 6.76 ± 0.06 | −0.35 ± 0.19 | 6.22 ± 0.09 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Synthetic Procedures
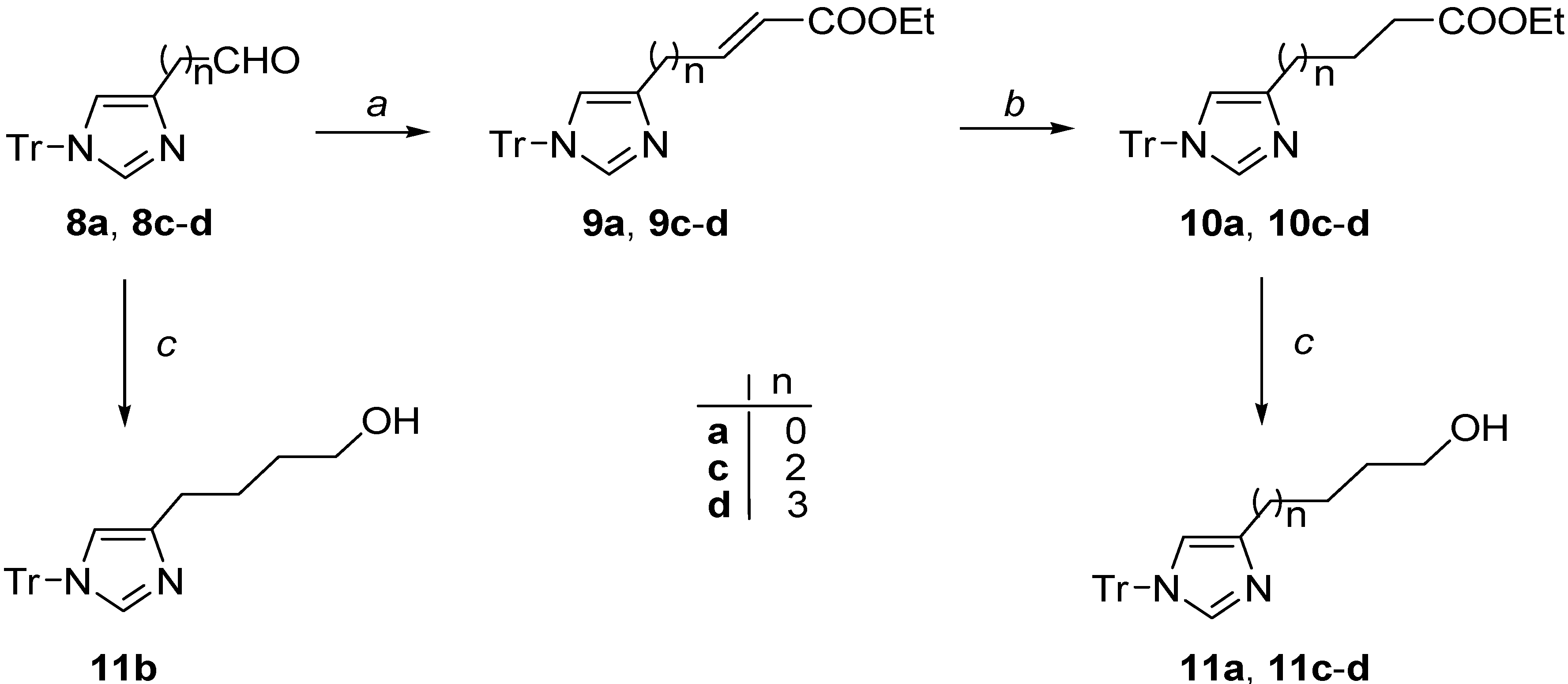
3.2.1. Synthesis of Unsaturated Esters 9a, 9c–d
3.2.2. (E)-Ethyl 3-(1-trityl-1H-imidazol-4-yl)acrylate (9a)
3.2.3. (E)-Ethyl 5-(1-trityl-1H-imidazol-4-yl)pent-2-enoate (9c)
3.2.4. (E-Ethyl 6-(1-trityl-1H-imidazol-4-yl)hex-2-enoate (9d)
3.2.5. Synthesis of Ester Derivatives 10a, 10c–d
3.2.6. Ethyl 3-(1-trityl-1H-imidazol-4-yl)propanoate (10a)
3.2.7. Ethyl 5-(1-trityl-1H-imidazol-4-yl)pentanoate (10c)
3.2.8. Ethyl 6-(1-trityl-1H-imidazol-4-yl)hexanoate (10d)
3.2.9. Synthesis of Alcohol Derivatives 11a, 11c–d
3.2.10. 3-(1-Trityl-1H-imidazol-4-yl)propan-1-ol (11a)
3.2.11. 4-(1-Trityl-1H-imidazol-4-yl)butan-1-ol (11b)
3.2.12. 5-(1-Trityl-1H-imidazol-4-yl)pentan-1-ol (11c)
3.2.13. 6-(1-Trityl-1H-imidazol-4-yl)hexan-1-ol (11d)
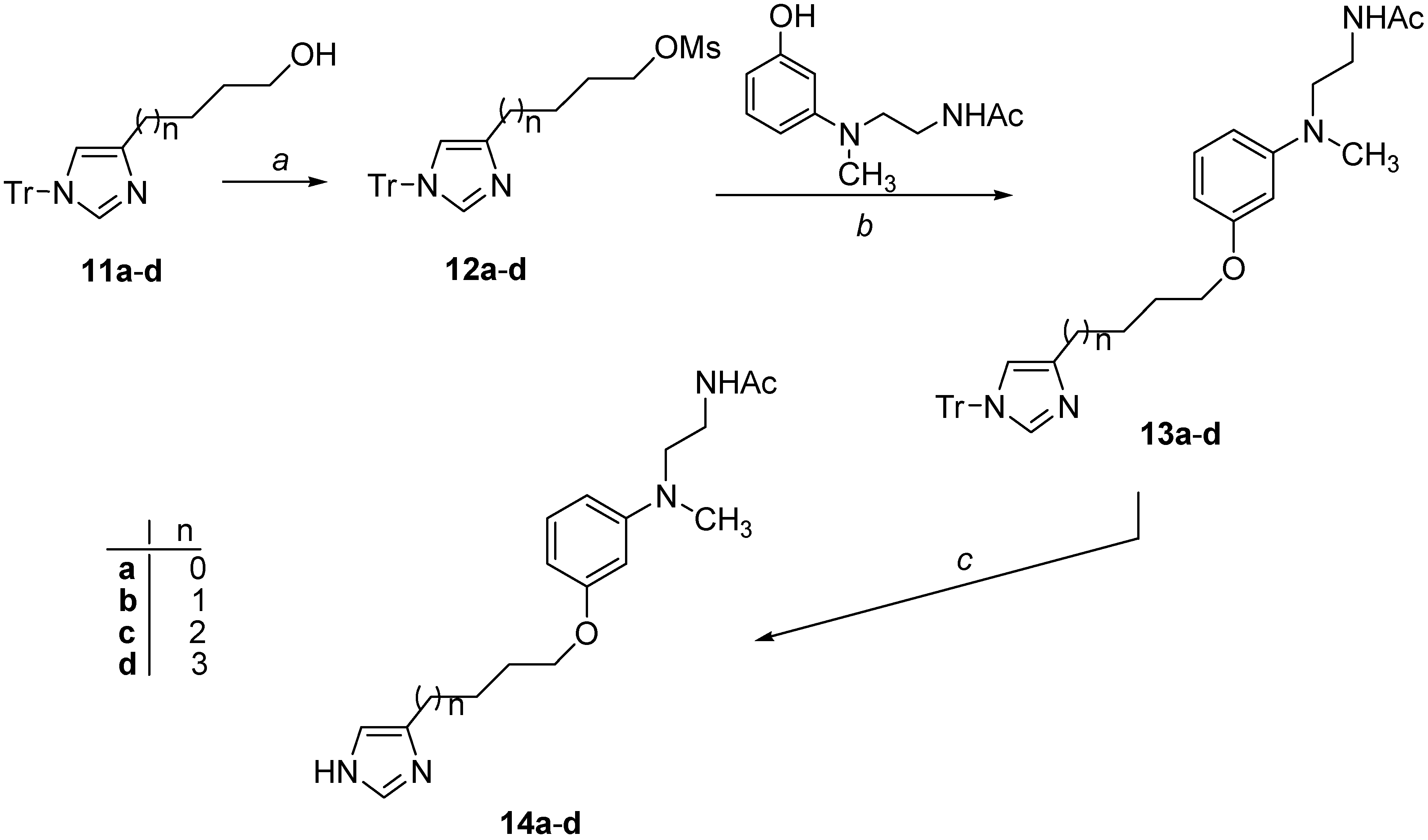
3.2.14. Synthesis of Mesyl Derivatives 12a–d
3.2.15. 3-(1-Trityl-1H-imidazol-4-yl)propyl methansulfonate (12a)
3.2.16. 4-(1-Trityl-1H-imidazol-4-yl)butyl methansulfonate (12b)
3.2.17. 5-(1-Trityl-1H-imidazol-4-yl)pentyl methansulfonate (12c)
3.2.18. 6-(1-Trityl-1H-imidazol-4-yl)hexyl methansulfonate (12d)
3.2.19. Synthesis of Derivatives 13a–d
3.2.20. N-[2-(Methyl{3-[3-(1-trityl-1H-imidazol-4-yl)propoxy]phenyl}amino)ethyl]acetamide (13a)
3.2.21. N-[2-(Methyl{3-[4-(1-trityl-1H-imidazol-4-yl)butoxy]phenyl}amino)ethyl]acetamide (13b)
3.2.22. N-[2-(Methyl{3-[5-(1-trityl-1H-imidazol-4-yl)pentyloxy]phenyl}amino)ethyl]acetamide (13c)
3.2.23. N-[2-(Methyl{3-[6-(1-trityl-1H-imidazol-4-yl)hexyloxy]phenyl}amino)ethyl]acetamide (13d)
3.2.24. Synthesis of Target Derivatives 14a–d
3.2.25. N-[2-({3-[3-(1H-Imidazol-4-yl)propoxy]phenyl}methylamino)ethyl]acetamide (14a)
3.2.26. N-[2-({3-[4-(1H-Imidazol-4-yl)butoxy]phenyl}methylamino)ethyl]acetamide (14b)
3.2.27. N-[2-({3-[5-(1H-Imidazol-4-yl)pentyloxy]phenyl}methylamino)ethyl]acetamide (14c)
3.2.28. N-[2-({3-[6-(1H-Imidazol-4-yl)hexyloxy]phenyl}methylamino)ethyl]acetamide (14d)
3.3. Human Histamine H3 Receptor Binding Assay
3.4. Human Melatonin Receptors Binding Assay
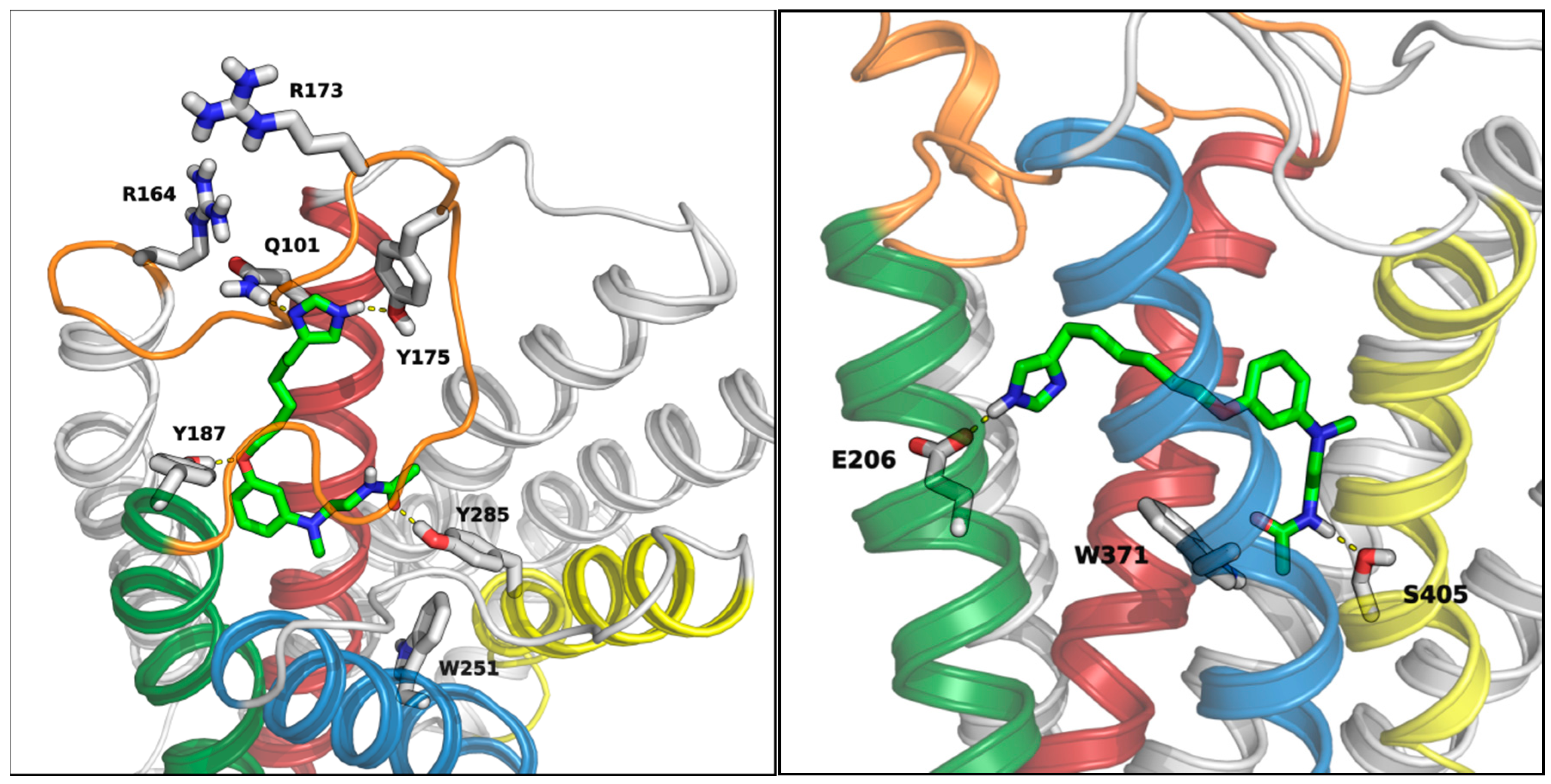
3.5. Docking Studies
3.5.1. MT1 Receptor
3.5.2. H3 Receptor
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Dubocovich, M.L.; Delagrange, P.; Krause, D.N.; Sugden, D.; Cardinali, D.P.; Olcese, J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol. Rev. 2010, 62, 343–380. [Google Scholar] [CrossRef]
- Slominski, R.M.; Reiter, R.J.; Schlabritz-Loutsevitch, N.; Ostrom, R.S.; Slominski, A.T. Melatonin membrane receptors in peripheral tissues: Distribution and functions. Mol. Cell Endocrinol. 2012, 35, 152–166. [Google Scholar]
- Zlotos, D.P.; Jockers, R.; Cecon, E.; Rivara, S.; Witt-Enderby, P.A. MT1 and MT2 melatonin receptors: Ligands, models, oligomers, and therapeutic potential. J. Med. Chem. 2014, 57, 3161–3185. [Google Scholar] [CrossRef]
- Pandi-Perumal, S.R.; Trakht, I.; Srinivasan, V.; Spence, D.W.; Maestroni, G.J.M.; Zisapel, N.; Cardinali, D.P. Physiological effects of melatonin: Role of melatonin receptors and signal transduction pathways. Prog. Neurobiol. 2008, 85, 335–353. [Google Scholar] [CrossRef]
- Kim, T.K.; Kleszczynski, K.; Janjetovic, Z.; Sweatman, T.; Lin, Z.; Li, W.; Reiter, R.J.; Fischer, T.W.; Slominski, A.T. Metabolism of melatonin and biological activity of intermediates of melatoninergic pathway in human skin cells. FASEB J. 2013, 27, 2742–2755. [Google Scholar] [CrossRef]
- Cajochen, C.; Kräuchi, K.; Wirz-Justice, A. Role of melatonin in the regulation of human circadian rhythms and sleep. J. Neuroendocrinol. 2003, 15, 432–437. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Mayo, J.C.; Sainz, R.M.; Leon, J.; Czarnocki, Z. Melatonin as an antioxidant: Biochemical mechanisms and pathophysiological implications in humans. Acta Biochim. Pol. 2003, 50, 1129–1146. [Google Scholar]
- Srinivasan, V.; Pandi-Perumal, S.R.; Cardinali, D.P.; Poeggeler, B.; Hardeland, R. Melatonin in Alzheimer’s disease and other neurodegenerative disorders. Behav. Brain Funct. BBF 2006, 2, 15. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z. Role of melatonin in Alzheimer-like neurodegeneration. Acta Pharmacol. Sin. 2006, 27, 41–49. [Google Scholar] [CrossRef]
- Wang, X. The antiapoptotic activity of melatonin in neurodegenerative diseases. CNS Neurosci. Ther. 2009, 15, 345–357. [Google Scholar] [CrossRef]
- Das, A.; McDowell, M.; Pava, M.J.; Smith, J.A.; Reiter, R.J.; Woodward, J.J.; Varma, A.K.; Ray, S.K.; Banik, N.L. The inhibition of apoptosis by melatonin in VSC4.1 motoneurons exposed to oxidative stress, glutamate excitotoxicity, or TNF-alpha toxicity involves membrane melatonin receptors. J. Pineal Res. 2010, 48, 157–169. [Google Scholar] [CrossRef]
- Das, A.; Wallace, G.; Reiter, R.J.; Varma, A.K.; Ray, S.K.; Banik, N.L. Overexpression of melatonin membrane receptors increases calcium-binding proteins and protects VSC4.1 motoneurons from glutamate toxicity through multiple mechanisms. J. Pineal Res. 2013, 54, 58–68. [Google Scholar]
- Chern, C.-M.; Liao, J.-F.; Wang, Y.-H.; Shen, Y.-C. Melatonin ameliorates neural function by promoting endogenous neurogenesis through the MT2 melatonin receptor in ischemic-stroke mice. Free Radic. Biol. Med. 2012, 52, 1634–1647. [Google Scholar] [CrossRef]
- Kaneko, Y.; Hayashi, T.; Yu, S.; Tajiri, N.; Bae, E.C.; Solomita, M.A.; Chheda, S.H.; Weinbren, N.L.; Parolini, O.; Borlongan, C.V. Human amniotic epithelial cells express melatonin receptor MT1, but not melatonin receptor MT2: A new perspective to neuroprotection. J. Pineal Res. 2011, 50, 272–280. [Google Scholar] [CrossRef]
- Carocci, A.; Catalano, A.; Bruno, C.; Lovece, A.; Roselli, M.G.; Cavalluzzi, M.M.; de Santis, F.; de Palma, A.; Rusciano, M.R.; Illario, M.; et al. N-(phenoxyalkyl)amides as M1 and MT2 ligands: Antioxidant properties and inhibition of Ca2+/CaM-dependent kinase II. Bioorg. Med. Chem. 2013, 21, 847–851. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sirianni, A.; Pei, Z.; Cormier, K.; Smith, K.; Jiang, J.; Zhou, S.; Wang, H.; Zhao, R.; Yano, H.; et al. The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity. J. Neurosci. 2011, 31, 14496–14507. [Google Scholar] [CrossRef]
- Rodríguez-Franco, M.I.; Fernández-Bachiller, M.I.; Pérez, C.; Hernández-Ledesma, B.; Bartolomé, B. Novel tacrine-melatonin hybrids as dual-acting drugs for Alzheimer disease, with improved acetylcholinesterase inhibitory and antioxidant properties. J. Med. Chem. 2006, 49, 459–462. [Google Scholar] [CrossRef]
- López-Iglesias, B.; Pérez, C.; Morales-García, J.A.; Alonso-Gil, S.; Pérez-Castillo, A.; Romero, A.; López, M.G.; Villarroya, M.; Conde, S.; Rodríguez-Franco, M.I. New melatonin-N,N-dibenzyl(N-methyl)amine hybrids: Potent neurogenic agents with antioxidant, cholinergic, and neuroprotective properties as innovative drugs for Alzheimer’s disease. J. Med. Chem. 2014, 57, 3773–3785. [Google Scholar] [CrossRef]
- Chojnacki, J.E.; Liu, K.; Yan, X.; Toldo, S.; Selden, T.; Estrada, M.; Rodríguez-Franco, M.I.; Halquist, M.S.; Ye, D.; Zhang, S. Discovery of 5-(4-Hydroxyphenyl)-3-oxo-pentanoic acid [2-(5-methoxy-1H-indol-3-yl)-ethyl]-amide as a neuroprotectant for Alzheimer’s disease by hybridization of curcumin and melatonin. ACS Chem. Neurosci. 2014, 5, 690–699. [Google Scholar] [CrossRef]
- Leurs, R.; Bakker, R.A.; Timmerman, H.; de Esch, I.J. The histamine H3 receptor: From gene cloning to H3 receptor drugs. Nat. Rev. Drug Discov. 2005, 4, 107–120. [Google Scholar] [CrossRef]
- Singh, M.; Jadhav, H.R. Histamine H3 receptor function and ligands: Recent developments. Mini-Rev. Med. Chem. 2013, 12, 47–57. [Google Scholar] [CrossRef]
- Gemkow, M.J.; Davenport, A.J.; Harich, S.; Ellenbroek, B.A.; Cesura, A.; Hallett, D. The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov. Today 2009, 14, 509–515. [Google Scholar] [CrossRef]
- Brioni, J.D.; Esbenshade, T.A.; Garrison, T.R.; Bitner, S.R.; Cowart, M.D. Discovery of histamine H3 antagonists for the treatment of cognitive disorders and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2011, 336, 38–46. [Google Scholar] [CrossRef]
- Bhowmik, M.; Khanam, R.; Vohora, D. Histamine H3 receptor antagonists in relation to epilepsy and neurodegeneration: A systemic consideration of recent progress and perspectives. Br. J. Pharmacol. 2012, 167, 1398–1414. [Google Scholar] [CrossRef]
- Bitner, R.S.; Markosyan, S.; Nikkel, A.L.; Brioni, J.D. In-vivo histamine H3 receptor antagonism activates cellular signaling suggestive of symptomatic and disease modifying efficacy in Alzheimer’s disease. Neuropharmacology 2011, 60, 460–466. [Google Scholar] [CrossRef]
- Yan, H.; Zhang, X.; Hu, W.; Ma, J.; Hou, W.; Zhang, X.; Wang, X.; Gao, J.; Shen, Y.; Lv, J.; et al. Histamine H3 receptors aggravate cerebral ischaemic injury by histamine-independent mechanisms. Nat. Commun. 2014, 5, 3334. [Google Scholar]
- Bhowmik, M.; Saini, N.; Vohora, D. Histamine H3 receptor antagonism by ABT-239 attenuates kainic acid induced excitotoxicity in mice. Brain Res. 2014. [Google Scholar] [CrossRef]
- Tang, L.; Zhao, L.; Hong, L.; Yang, F.; Sheng, R.; Chen, J.; Shi, Y.; Zhou, N.; Hu, Y. Design and synthesis of novel 3-substituted-indole derivatives as selective H3 receptor antagonists and potent free radical scavengers. Bioorg. Med. Chem. 2013, 21, 5936–5944. [Google Scholar] [CrossRef]
- Mésangeau, C.; Pérès, B.; Descamps-François, C.; Chavatte, P.; Audinot, V.; Coumailleau, S.; Boutin, J.A.; Delagrange, P.; Bennejean, C.; Renard, P.; et al. Design, synthesis and pharmacological evaluation of novel naphthalenic derivatives as selective MT1 melatoninergic ligands. Bioorg. Med. Chem. 2010, 18, 3426–3436. [Google Scholar] [CrossRef]
- Markl, C.; Clafshenkel, W.P.; Attia, M.I.; Sethi, S.; Witt-Enderby, P.A.; Zlotos, D.P. N-acetyl-5-arylalkyloxytryptamine analogs: Probing the melatonin receptors for MT1-selectivity. Arch. Pharm. (Weinheim) 2011, 344, 666–674. [Google Scholar] [CrossRef]
- Rivara, S.; Pala, D.; Lodola, A.; Mor, M.; Lucini, V.; Dugnani, S.; Scaglione, F.; Bedini, A.; Lucarini, S.; Tarzia, G.; et al. MT1-selective melatonin receptor ligands: Synthesis, pharmacological evaluation, and molecular dynamics investigation of N-{[(3-O-substituted)anilino]alkyl}amides. ChemMedChem 2012, 7, 1954–1964. [Google Scholar] [CrossRef]
- Celanire, S.; Wijtmans, M.; Talaga, P.; Leurs, R.; de Esch, I.J.P. Histamine H3 receptor antagonists reach out for the clinic. Drug Discov. Today 2005, 10, 1613–1627. [Google Scholar] [CrossRef]
- Wingen, K.; Stark, H. Scaffold variations in amine warhead of histamine H3 receptor antagonists. Drug Discov. Today Technol. 2013, 10, e483–e489. [Google Scholar] [CrossRef]
- Tedford, C.E.; Hoffmann, M.; Seyedi, N.; Maruyama, R.; Levi, R.; Yates, S.L.; Ali, S.M.; Phillips, J.G. High antagonist potency of GT-2227 and GT-2331, new histamine H3 receptor antagonists, in two functional models. Eur. J. Pharmacol. 1998, 351, 307–311. [Google Scholar] [CrossRef]
- De Esch, I.J.; Gaffar, A.; Menge, W.M.; Timmerman, H. Synthesis and histamine H3 receptor activity of 4-(n-alkyl)-1H-imidazoles and 4-(ω-phenylalkyl)-1H-imidazoles. Bioorg. Med. Chem. 1999, 7, 3003–3009. [Google Scholar] [CrossRef]
- Vollinga, R.C.; Menge, W.M.; Leurs, R.; Timmerman, H. New analogs of burimamide as potent and selective histamine H3 receptor antagonists: The effect of chain length variation of the alkyl spacer and modifications of the N-thiourea substituent. J. Med. Chem. 1995, 38, 2244–2250. [Google Scholar] [CrossRef]
- Tozer, M.J.; Harper, E.A.; Kalindjian, S.B.; Pether, M.J.; Shankley, N.P.; Watt, G.F. From histamine to imidazolylalkyl-sulfonamides: The design of a novel series of histamine H3-receptor antagonists. Bioorg. Med. Chem. Lett. 1999, 9, 1825–1830. [Google Scholar] [CrossRef]
- Grassmann, S.; Apelt, J.; Sippl, W.; Ligneau, X.; Pertz, H.H.; Zhao, Y.H.; Arrang, J.M.; Ganellin, C.R.; Schwartz, J.C.; Schunack, W.; et al. Imidazole derivatives as a novel class of hybrid compounds with inhibitory histamine N-methyltransferase potencies and histamine hH3 receptor affinities. Bioorg. Med. Chem. 2003, 11, 2163–2174. [Google Scholar] [CrossRef]
- Ettaoussi, M.; Sabaouni, A.; Rami, M.; Boutin, J.A.; Delagrange, P.; Renard, P.; Spedding, M.; Caignard, D.H.; Berthelot, P.; Yous, S. Design, synthesis and pharmacological evaluation of new series of naphthalenic analogues as melatoninergic (MT1/MT2) and serotoninergic 5-HT2C dual ligands (I). Eur. J. Med. Chem. 2012, 49, 310–323. [Google Scholar] [CrossRef]
- Zlotos, D.P.; Attia, M.I.; Julius, J.; Sethi, S.; Witt-Enderby, P.A. 2-[(2,3-dihydro-1H-indol-1-yl)methyl]melatonin analogues: A novel class of MT2-selective melatonin receptor antagonists. J. Med. Chem. 2009, 52, 826–833. [Google Scholar] [CrossRef]
- Rivara, S.; Lodola, A.; Mor, M.; Bedini, A.; Spadoni, G.; Lucini, V.; Pannacci, M.; Fraschini, F.; Scaglione, F.; Sanchez, R.O.; et al. N-(Substituted-anilinoethyl)amides: Design, synthesis, and pharmacological characterization of a new class of melatonin receptor ligands. J. Med. Chem. 2007, 50, 6618–6626. [Google Scholar] [CrossRef]
- Stark, H.; Ligneau, X.; Sadek, B.; Ganellin, C.R.; Arrang, J.M.; Schwartz, J.C.; Schunack, W. Analogues and derivatives of ciproxifan, a novel prototype for generating potent histamine H3-receptor antagonists. Bioorg. Med. Chem. Lett. 2000, 10, 2379–2382. [Google Scholar] [CrossRef]
- Uveges, A.J.; Kowal, D.; Zhang, Y.; Spangler, T.B.; Dunlop, J.; Semus, S.; Jones, P.G. The role of transmembrane helix 5 in agonist binding to the human H3 receptor. J. Pharmacol. Exp. Ther. 2002, 301, 451–458. [Google Scholar] [CrossRef]
- Yao, B.B.; Hutchins, C.W.; Carr, T.L.; Cassar, S.; Masters, J.N.; Bennani, J.L.; Esbenshade, T.A.; Hancock, A.A. Molecular modeling and pharmacological analysis of spacies-related histamine H3 receptor heterogeneity. Neuropharmacology 2003, 44, 773–786. [Google Scholar] [CrossRef]
- Bordi, F.; Rivara, S.; Dallaturca, E.; Carmi, C.; Pala, D.; Lodola, A.; Vacondio, F.; Flammini, L.; Bertoni, S.; Ballabeni, V.; et al. Dibasic biphenyl H3 receptor antagonists: Steric tolerance for a lipophilic side chain. Eur. J. Med. Chem. 2012, 48, 214–230. [Google Scholar] [CrossRef]
- Ly, K.S.; Letavic, M.A.; Keith, J.M.; Miller, J.M.; Stocking, E.M.; Barbier, A.J.; Bonaventure, P.; Lord, B.; Jiang, X.; Boggs, J.D.; et al. Synthesis and biological activity of piperazine and diazepane amides that are histamine H3 antagonists and serotonin reuptake inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 39–43. [Google Scholar] [CrossRef]
- Roche, O.; Rodriguez Sarmiento, R.M. A new class of histamine H3 receptor antagonists derived from ligand based design. Bioorg. Med. Chem. Lett. 2007, 17, 3670–3675. [Google Scholar] [CrossRef]
- Kitbunnadaj, R.; Hoffmann, M.; Fratantoni, S.A.; Bongers, G.; Bakker, R.A.; Wieland, K.; el Jilali, A.; de Esch, I.J.P.; Menge, W.M.P.B.; Timmerman, H.; et al. New high affinity H3 receptor agonists without a basic side chain. Bioorg. Med. Chem. 2005, 13, 6309–6323. [Google Scholar] [CrossRef]
- Griffith, R.K.; DiPietro, R.A. Improved syntheses of vinylimidazoles. Synth. Commun. 1986, 16, 1761–1770. [Google Scholar] [CrossRef]
- Lovenberg, T.W.; Roland, B.L.; Wilson, S.J.; Jiang, X.; Pyati, J.; Huvar, A.; Jackson, M.R.; Erlander, M.G. Cloning and functional expression of the human histamine H3 receptor. Mol. Pharmacol. 1999, 55, 1101–1107. [Google Scholar]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Nonno, R.; Lucini, V.; Pannacci, M.; Mazzucchelli, C.; Angeloni, D.; Fraschini, F.; Stankov, B.M. Pharmacological characterization of the human melatonin Mel8a receptor following stable transfection into NIH3T3 cells. Br. J. Pharmacol. 1998, 124, 485–492. [Google Scholar] [CrossRef]
- Nonno, R.; Pannacci, M.; Lucini, V.; Angeloni, D.; Fraschini, F.; Stankov, B.M. Ligand efficacy and potency at recombinant human MT2 melatonin receptors: Evidence for agonist activity of some mt1-antagonists. Br. J. Pharmacol. 1999, 127, 1288–1294. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Spadoni, G.; Balsamini, C.; Bedini, A.; Diamantini, G.; di Giacomo, B.; Tontini, A.; Tarzia, G.; Mor, M.; Plazzi, P.V.; Rivara, S.; et al. 2-[N-Acylamino(C1-C3)alkyl]indoles as MT1 melatonin receptor partial agonists, antagonists, and putative inverse agonists. J. Med. Chem. 1998, 41, 3624–3634. [Google Scholar] [CrossRef]
- Glide, version 5.7; Schrödinger, LLC:: New York, NY, USA, 2011.
- Maestro, version 9.2; Schrödinger, LLC: New York, NY, USA, 2011.
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pala, D.; Scalvini, L.; Lodola, A.; Mor, M.; Flammini, L.; Barocelli, E.; Lucini, V.; Scaglione, F.; Bartolucci, S.; Bedini, A.; et al. Synthesis and Characterization of New Bivalent Agents as Melatonin- and Histamine H3-Ligands. Int. J. Mol. Sci. 2014, 15, 16114-16133. https://doi.org/10.3390/ijms150916114
Pala D, Scalvini L, Lodola A, Mor M, Flammini L, Barocelli E, Lucini V, Scaglione F, Bartolucci S, Bedini A, et al. Synthesis and Characterization of New Bivalent Agents as Melatonin- and Histamine H3-Ligands. International Journal of Molecular Sciences. 2014; 15(9):16114-16133. https://doi.org/10.3390/ijms150916114
Chicago/Turabian StylePala, Daniele, Laura Scalvini, Alessio Lodola, Marco Mor, Lisa Flammini, Elisabetta Barocelli, Valeria Lucini, Francesco Scaglione, Silvia Bartolucci, Annalida Bedini, and et al. 2014. "Synthesis and Characterization of New Bivalent Agents as Melatonin- and Histamine H3-Ligands" International Journal of Molecular Sciences 15, no. 9: 16114-16133. https://doi.org/10.3390/ijms150916114